
Structure-based virtual screening and experimental validation of the discovery of inhibitors targeted towards the human coronavirus nucleocapsid protein†
Chung-ke
Chang‡
a,
Sivakamavalli
Jeyachandran‡
b,
Nien-Jen
Hu‡
c,
Chia-Ling
Liu‡
b,
Shing-Yen
Lin
b,
Yong-Sheng
Wang
b,
Yu-Ming
Chang
d and
Ming-Hon
Hou
*bc
aInstitute of Biomedical Science, Academia Sinica, Nangang, Taipei, Taiwan
bInstitute of Genomics and Bioinformatics and Institute of Life Sciences, National Chung Hsing University, Taichung 40254, Taiwan. E-mail: mhho@nchu.edu.tw; Fax: +886-4-2285-9329; Tel: +886-4-2284-0338 ext. 7011
cInstitute of Biochemistry, National Chung Hsing University, Taichung 40254, Taiwan
dInstitute of Biological Chemistry, Academia Sinica, Taipei 11529, Taiwan
First published on 20th October 2015
Abstract
Nucleocapsid protein (NP), an essential RNA-binding viral protein in human coronavirus (CoV)-infected cells, is required for the replication and transcription of viral RNA. Recent studies suggested that human CoV NP is a valid target for antiviral drug development. Based on this aspect, structure-based virtual screening targeting nucleocapsid protein (NP) was performed to identify good chemical starting points for medicinal chemistry. The present study utilized structure-based virtual screening against human CoV-OC43 using the Zinc database, which is performed through docking with varying precisions and computational intensities to identify eight potential compounds. The chosen potential leads were further validated experimentally using biophysical means. Surface plasmon resonance (SPR) analysis indicated that one among the potential leads, 6-chloro-7-(2-morpholin-4-yl-ethylamino) quinoxaline-5,8-dione (small-compound H3), exhibited a significant decrease of RNA-binding capacity of NP by more than 20%. The loss of binding activity was manifested as a 20% decrease in the minimum on-rate accompanied with a 70% increase in the maximum off-rate. Fluorescence titration and X-ray crystallography studies indicated that H3 antagonizes the binding between HCoV-OC43 NP and RNA by interacting with the N-terminal domain of the NP. Our findings provide insight into the development of new therapeutics that disrupt the interaction between RNA and viral NP in the HCoV. The discovery of the new compound would be an impetus to design novel NP inhibitors against human CoV.
Introduction
Respiratory tract infection (RTI) is one of the infectious diseases causing serious mortality among young children and adults in developing countries.1–3 Most of the RTI diseases are caused by RNA viruses, including respiratory syncytial virus (RSV), influenza viruses, rhinoviruses, parainfluenza viruses, human metapneumovirus and human coronaviruses (HCoVs).4–8 Coronaviruses (CoVs) are positive single-strand, enveloped RNA viruses with genome sizes ranging from 27 to 33 kilobases (kb) and belong to the Coronaviridae family. They are classified into α-, β-, γ- and δ-coronaviruses.9 HCoV-NL63 and HCoV-229E belong to the α-coronaviruses while HCoV-OC43, HCoV-HKU1, and the severe acute respiratory syndrome coronavirus (SARS-CoV) belong to the β-coronaviruses.9 The pathogen middle east respiratory syndrome coronavirus (MERS-CoV) belongs to the genus β-coronavirus.10Due to the severe pathogenic capability, e.g. severe acute respiratory syndrome (SARS) pandemic in Asia during 2003–2004 and the recent outbreak of middle-east respiratory syndrome (MERS), and their dreadful nature, human coronaviruses have attracted clinical interest, recently.11–13 Moreover, there are no efficacious therapies against coronaviral diseases so far, promoting the development of anti-coronavirus compounds. CoVs are composed of several structural proteins sharing high conservation across species: the small envelope (E) protein, matrix (M) glycoprotein, the trimeric spike (S) glycoprotein bound to the viral membrane, and the nucleocapsid protein (NP) residing inside the virus. Some variants contain a third membrane-bound glycoprotein, HE (hemagglutinin-esterase).14 The nucleocapsid protein (NP) is one of the most abundant structural proteins in CoVs, and serves multiple functions, such as ribonucleoprotein formation, modulation of host cell metabolism, and regulation of viral RNA synthesis during replication and transcription.15,16
In addition, coronavirus NPs consist of 3 domains: an N-terminal RNA-binding domain (NTD), an intrinsically disordered central Ser/Arg (SR)-rich linker that may contain the protein's primary phosphorylation sites, and a C-terminal dimerisation domain (CTD). Crystal structures of several coronavirus NP-NTDs have been determined previously, including those encoded by infectious bronchitis virus (IBV), mouse hepatitis virus (MHV), HCoV-OC43, and SARS-CoV.17–22 Furthermore, crystal structures of CoV NPs-CTDs provide structural clues on how the building blocks of coronavirus nucleocapsid form through the NP dimer.17–19,22–25 Because of their importance in the viral life cycle, NPs are becoming unique targets for therapeutic development against viral diseases.16 Two strategies to inhibit oligomeric NP function have been reported.26 The first is to enhance or inhibit its oligomerization properties.27 The second strategy is to disrupt the RNA-binding site.28 We have previously identified a unique ribonucleotide-binding pocket consisting of highly conserved residues in the centre of NTD by solving the complex structure of CoV NP-NTD bound with a ribonucleotide monophosphate. Compounds binding to this RNA-binding pocket may inhibit normal NP function and could be used to combat CoV diseases.28
Recently, this problematic research has attracted particular interest in virology due to the demand for new antivirals, preferably with a completely different mechanism of action from traditional antiviral drugs to avoid resistance and cross-resistance. The interaction between HCoV NP and RNA is an optimal target for CoV drug design. The strategy would be helpful to find out new potential leads to disrupt the functions of HCoV NP. In this paper we utilized the crystal structure of HCoV NP NTD27 as the target of a virtual screening endeavor using LIBDOCK. Eight potential leads (H1–H8) were initially selected targeting the active site of HCoV NPs. We further studied the effects of the eight compounds on the RNA-binding affinity of NP by SPR experiments. Among the 8 leads, 6-chloro-7-(2-morpholin-4-yl-ethylamino) quinoxaline-5,8-dione (H3) revealed inhibitory activity on the RNA-binding of NP. The crystal structure of HCoV-OC43 NP-NTD in complex with H3 was also determined depicting the detailed binding interactions. Our findings provide new insights into the development of novel drugs that may disrupt the interaction between RNA and viral NPs in HCoVs.
Materials and methods
Protein preparation
Expression and purification of the NTD of HCoV-OC43 NP were performed as previously described.27 The purified protein was concentrated using an Amicon Ultra-15 centrifugal filter unit (Millipore, MA, USA) with a molecular weight cut-off of 3 kDa and stored at −80 °C. The Bradford method was used to determine protein concentrations using Bio-Rad protein assay reagents (Bio-Rad, CA, USA).Structure-based virtual screening
Ligand screening and molecular docking were performed with the LIBDOCK module in Discovery Studio, version 2.5 (DS 2.5). The publicly available ZINC database version 12 was chosen for virtual screening. The database was initially filtered to remove unreasonable molecules with unwanted physical and chemical properties. Molecules with the following properties were removed by default: molecular weight >600, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P < −4, logP > 8, hydrogen bond donors and acceptors >12, rotatable bonds >10, polar surface area >140, single bond chain length >6, chiral centers >4, unconstrained chiral centers >3, transition metals >8 rings, and d-hybrids. After filtering, the selected molecules comprised of over 20000 compounds from several drug databanks in the ZINC database were utilized to generate multiple conformations using the conformation search and minimization program in DS 2.5. The final 3D multiple conformations database was subjected to the pharmacophore query. The LIBDOCK molecular docking software was used to screen for small molecules that may bind to a structure of the NTD of HCoV-OC43 NP using default parameters. The binding pocket of the NTD, which includes Tyr124, Tyr126, Arg122, and Arg164, was represented by a set of spheres during the docking process. It produces protein–ligand complexes and each conformer was simultaneously subjected to side chain and backbone refinements. The conformers were ranked within 30 kcal mol−1 of the minimum energy using the energy minimization module. The screened conformers were passed to a final round of docking and scoring. The non-bonding interactions between the inhibitors and the receptor proteins were displayed in Pymol. The 8 potential hits were identified by looking for interaction characteristics (e.g. π–π stacking interactions) between HCoV-OC43 NP-NTD and hit compounds which closely mimic the ones between HCoV-OC43 NP-NTD and ribonucleotide.
P < −4, logP > 8, hydrogen bond donors and acceptors >12, rotatable bonds >10, polar surface area >140, single bond chain length >6, chiral centers >4, unconstrained chiral centers >3, transition metals >8 rings, and d-hybrids. After filtering, the selected molecules comprised of over 20000 compounds from several drug databanks in the ZINC database were utilized to generate multiple conformations using the conformation search and minimization program in DS 2.5. The final 3D multiple conformations database was subjected to the pharmacophore query. The LIBDOCK molecular docking software was used to screen for small molecules that may bind to a structure of the NTD of HCoV-OC43 NP using default parameters. The binding pocket of the NTD, which includes Tyr124, Tyr126, Arg122, and Arg164, was represented by a set of spheres during the docking process. It produces protein–ligand complexes and each conformer was simultaneously subjected to side chain and backbone refinements. The conformers were ranked within 30 kcal mol−1 of the minimum energy using the energy minimization module. The screened conformers were passed to a final round of docking and scoring. The non-bonding interactions between the inhibitors and the receptor proteins were displayed in Pymol. The 8 potential hits were identified by looking for interaction characteristics (e.g. π–π stacking interactions) between HCoV-OC43 NP-NTD and hit compounds which closely mimic the ones between HCoV-OC43 NP-NTD and ribonucleotide.
SDS-PAGE analysis
Polyacrylamide gel electrophoresis (12%) was performed on eluted fractions under reducing (β-mercaptoethanol) and non-reducing conditions.29 After electrophoresis, the gel was stained with Coomassie brilliant blue (BioShop, Taiwan). The molecular mass of the purified HCoV-OC43 NP-NTD was determined by comparison of its electrophoretic mobility with that of molecular mass marker proteins (GeneDireX, Taiwan). The homogeneity and molecular mass of the HCoV-OC43 NP-NTD were also confirmed by gel filtration on a FPLC column (GE Healthcare Bio-Sciences, Taiwan). Total protein concentration was determined by the Bradford method21 using bovine serum albumin (BSA) as a standard.Crystallization and data collection
Crystals of HCoV-OC43 NP-NTD-H3 complexes were obtained by co-crystallization using an HCoV-OC43 NP-NTD solution (8 mg ml−1) pre-incubated for 30 min with H3. High-quality crystals suitable for X-ray diffraction were grown by standing a reservoir solution containing 30% (v/v) glycerol as cryo-protectant at room temperature for 2 days.27 Crystals were flash cooled under a nitrogen-gas stream at 100 K. X-ray diffraction data for the HCoV-OC43 NP-NTD were collected at the BL13B1 beam line of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan).28 All diffraction images were recorded using an ADSD Q315 CCD detector, and the data were processed and scaled using the HKL2000 software package.30 Data collection statistics are summarized in Table 2.Structure determination and refinement
Because the new crystals grown in this study were isomorphous to those from HCoV-OC43 NP-NTD solved previously, the structures of the HCoV-OC43 NP-NTD complexes were determined using the structure of HCoV-OC43 NP-NTD (PDB ID: 3V3P) as a base.27 For each structure, iterative cycles of model building with Mifit and computational refinement with CNS were performed; 5% reflections were set aside for Rfree calculations.28,30 The stereochemical quality of the structures was assessed with the program PROCHECK.28 The molecular figures were produced with PyMOL (Shroedinger LLC, http://www.pymol.org).Surface plasmon resonance (SPR) binding experiments
A BIAcore 3000A SPR instrument (Pharmacia, Uppsala, Sweden) equipped with a SA5 sensor chip (Pharmacia) was used to obtain the association and dissociation rate constants between HCoV-OC43 NPs and RNA. The repeated intergenic sequence of HCoV-OC43, 5′-biotin-(UCUAAAC)4-3′, was used as a probe in our SPR experiments. Experiments were conducted by injecting NP in 50 mM tris (pH 7.5) with 150 mM NaCl and 0.1% CHAPS in the presence and absence of the H3 compound. The sensorgrams were fit to the 1:1 Langmuir model using the BIA evaluation software (version 3) to determine the association and dissociation rate constants (ka, kd).31 Binding affinities were calculated from the rate constants within the software. The chip surface was then washed with 10 μl of 10 mM HCl to eliminate non-specific binding. The second flow cell was unmodified and served as a control. A blank buffer solution was then passed over the chip to initiate the dissociation reaction; this step was continued for an additional 600 s until the reaction was complete. After 600 s, the surface was recovered by washing with 10 μl of 0.1% SDS for each single-stranded RNA. Before fitting to the 1:1 Langmuir model, the binding data were corrected by subtracting the control to account for refractive index differences.
Fluorescence spectroscopy
Experiments were conducted in buffer consisting of 50 mM tris (pH 7.3), 100 mM NaCl, and 0.1% CHAPS. Tryptophan fluorescence was measured at 330 nm with a Hitachi F-4500 fluorescence spectrophotometer using an excitation wavelength of 288 nm and a 1 cm light path. Emission data were also collected for wavelengths ranging from 300 to 400 nm. Three repeats were recorded for all static measurements. The relative fluorescence change intensity was determined using the following equation: change % = (FLNP − FLNP–drug)/FLNP × 100, where FLNP is the NP fluorescence in the absence of compound and FLNP–drug is the fluorescence of the NP–compound complex.32 In the fluorescence titration assay, a final concentration of 4 μM HCoV-OC43 NP was added to different concentrations of compound, and the samples were incubated at 4 °C for various durations. The binding constant between H3 and NP was obtained by fitting the average of H3-induced fluorescence changes (ΔF) from three separate experiments at 3 h following the addition of test compounds as previously described.28Results and discussion
The effects of 6-chloro-7-(2-morpholin-4-yl-ethylamino) quinoxaline-5,8-dione (H3) on the RNA binding activity of NP
Several potential hits, H1–H8, were identified with a high docking score for the distinct ribonucleotide-binding pocket with conserved residues (Arg 122, Tyr 124, Tyr 126, and Arg 164) (Fig. 1A and B), using a molecular docking software (LIBDOCK) described in the Materials and methods section. The RNA-binding capacities of HCoV-OC43 NP-NTD in the presence of these eight compounds were further studied by surface plasmon resonance (SPR). Only treatment with 6-chloro-7-(2-morpholin-4-yl-ethylamino) quinoxaline-5,8-dione (compound H3, Table S2, ESI,† and Fig. 2A) produced a significant decrease in the RNA-binding capacity of NP by more than 20%. Interestingly, H3 is a potent inhibitor of CDC25 protein phosphatases, exhibiting marked anti-proliferative activity against human MCF-7 breast cancer cells29 and arrest of synchronized cells in both the G1 and G2/M phases.19 Adding H3 in the assay resulted in a loss of binding capacity of NP to the RNA as evidenced by the decrease in response units (RU; Fig. 2B), suggesting that H3 hinders the binding of HCoV-OC43 NP to RNA. The loss of binding activity manifested as a 20% decrease in the minimum on-rate accompanied by a 70% increase in the maximum off-rate (Fig. 2C and Table 1).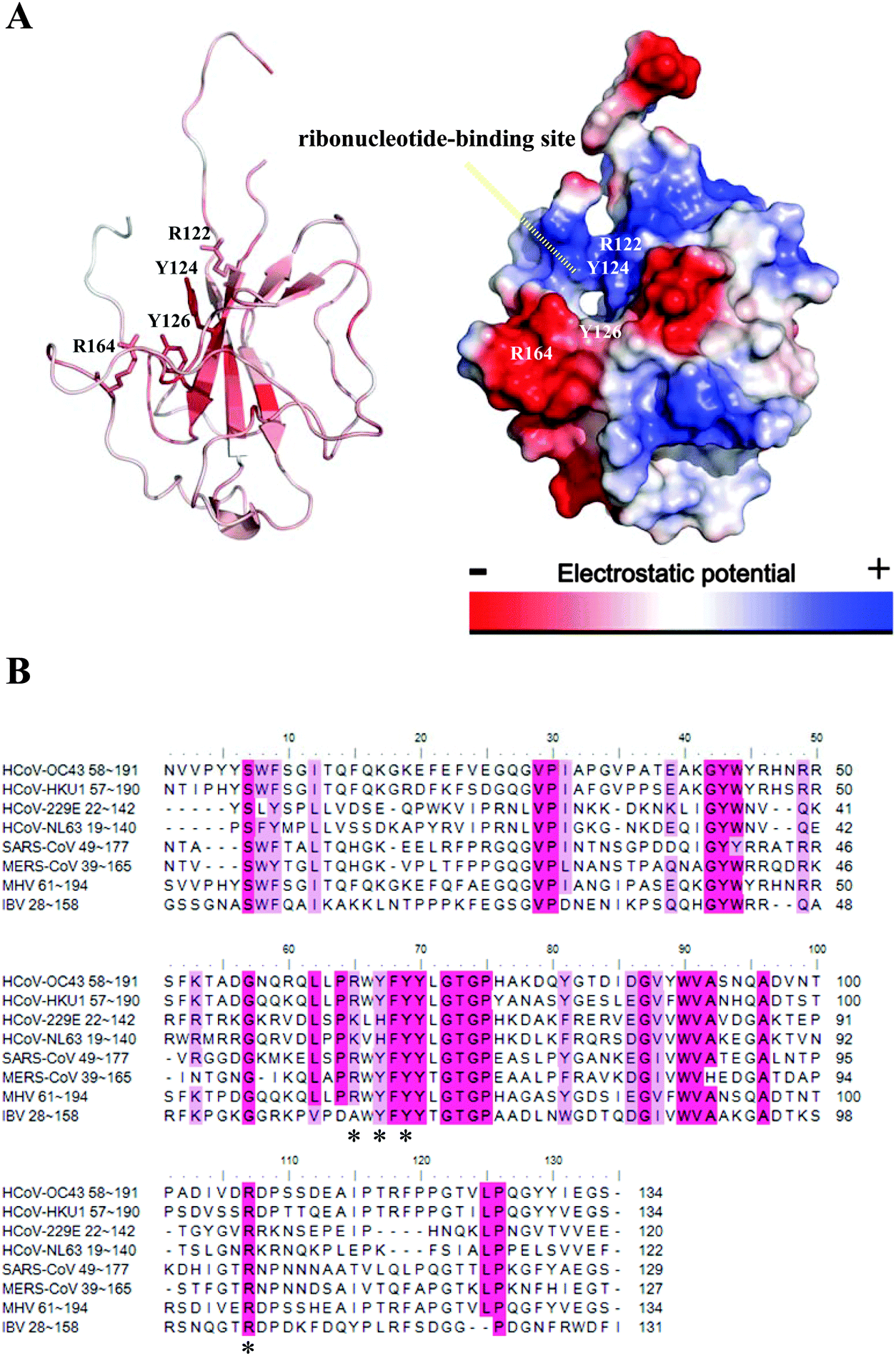 | ||
| Fig. 1 (A) Crystal structure of the HCoV-OC43 NP-NTD. The four conserved residues including Arg122, Tyr124, Tyr126, and Arg164 that contribute to the RNA binding at NP-NTD are marked (left). Surface representation of the HCoV-OC43 NP-NTD: electrostatic potentials are colored in blue (positive) and red (negative) (right). (B) Amino acid sequence alignment, performed using T-coffee, of N-NTDs from HCoV-OC43, HCoV-HKU1, HCoV-229E, HcoV-NL63, SARS-CoV, MERS, IBV and MHV, all of which were retrieved from GenBank. Conserved residues including Arg122, Tyr124, Tyr126, and Arg164 are indicated with an asterisk. | ||
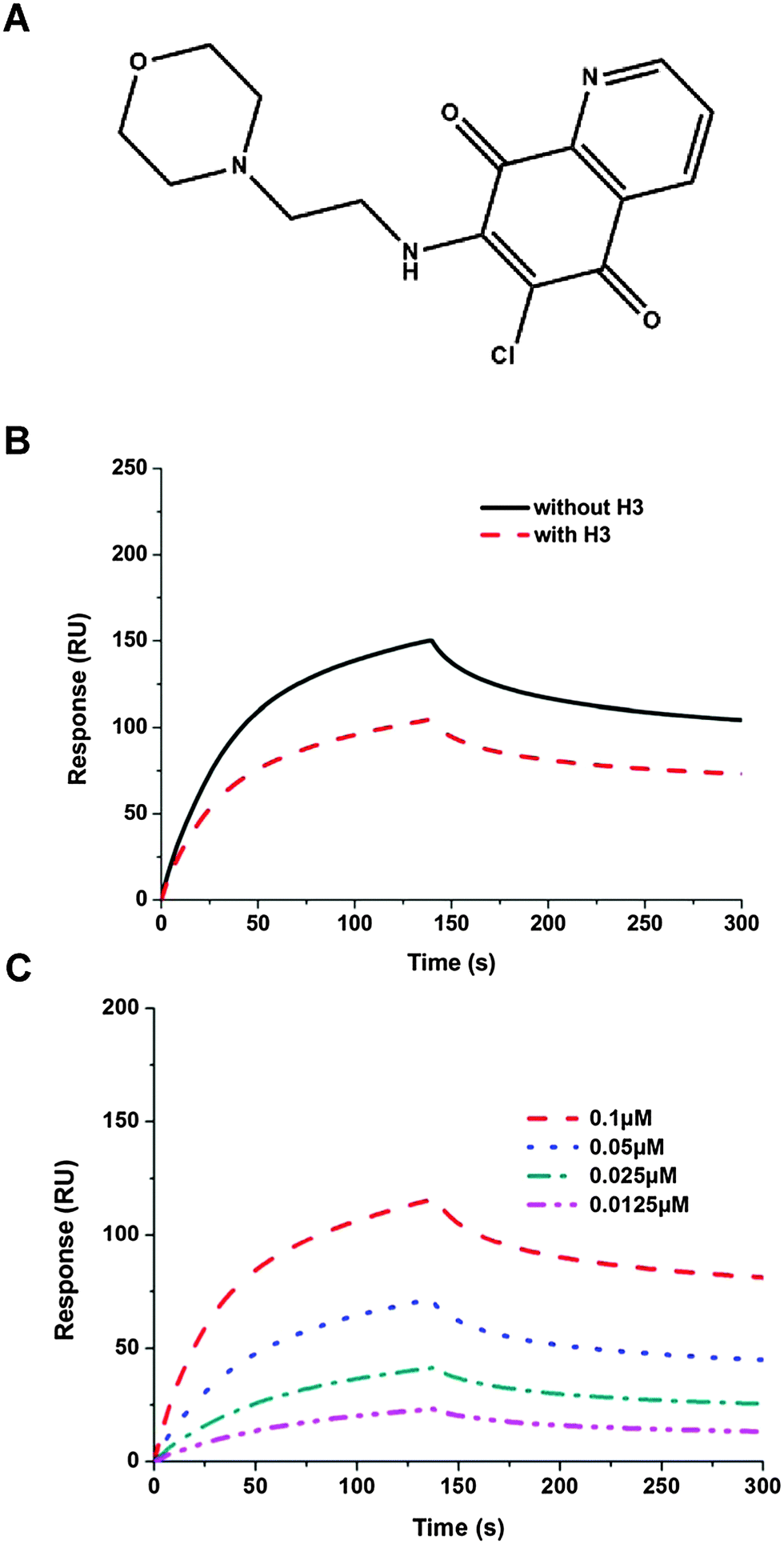 | ||
| Fig. 2 (A) Chemical structure of H3 (NSC663284). (B) Sensorgram of the interaction between the immobilized single-stranded RNA and full-length HCoV-OC43 NPs in the presence of H3 at 2 μM. (C) Sensorgram of the interaction between the immobilized single-stranded RNA and full-length HCoV-OC43 NPs at various concentrations in the presence of H3. | ||
| Sample | k d (S−1 M−1 × 10−4) | k a (S−1 × 104) | k D (nM) | BC (RU) |
|---|---|---|---|---|
| NP | 5.56 | 4.81 | 11.55 | 150 |
| NP + H3 | 6.49 | 3.76 | 17.3 | 116 |
| Crystal | HCoV-OC43 NP-NTD-H3 |
|---|---|
| a Values in parentheses are for the highest resolution shells. | |
| Data collection | NSRRC BL13B1 |
| Space group | P65 |
| Resolution (Å) | 30–2.50 (2.59–2.50)a |
| Wavelength (Å) | 1.00000 |
| Unit cell dimensions | |
| a = b (Å) | 81.931 |
| c (Å) | 42.853 |
| No. of reflections | |
| Observed | 47726 (4777) |
| Unique | 5733 (562) |
| Completeness (%) | 98.7 (100)a |
| R merge (%) | 2.4 (4.4)a |
| I/σ(I) | 83.1 (48.3)a |
| Refinement | |
| No. of reflections | 5707 (557) |
| R work (95% data) | 21.5 (24.8) |
| R free (5% data) | 26.9 (32.6) |
| Geometry deviations | |
| Bond lengths (Å) | 0.017 |
| Bond angles (°) | 2.0 |
| No. of all protein atoms | 1100 |
| Mean B-values (Å2) | 28.0 |
| No. of ligand atoms | 23 |
| Mean B-values (Å2) | 52.1 |
| No. of water molecules | 37 |
| Mean B-values (Å2) | 27.2 |
| Ramachandran plot (%) | |
| Most favored | 86.1 |
| Additionally allowed | 7.4 |
| Other | 6.5 |
We have shown that H3 interferes with the binding activity between HCoV-OC43 NPs and RNA, raising the possibility that H3 may also interact directly with the HCoV-OC43 NP. Thus, we tested this possibility using a fluorescence titration assay by scanning the fluorescence emission spectra of NP between 300 and 400 nm in the presence and absence of 2 μM H3 (Fig. 3A). The wavelength with maximal emission was determined to be at approximately 331 nm in both the presence and absence of H3, indicating that the interaction between H3 and NP did not noticeably shift the emission spectrum. To exclude the possibility that the change of NP fluorescence caused by H3 was due to an inner filter effect, we measured the absorption spectra from 330 to 400 nm for H3 at different concentrations and no significant absorbance was observed up to 5 μM H3 (Fig. S1, ESI†). Intriguingly, fluorescence titration assay showed that increasing drug concentration led to quenching of NP fluorescence (Fig. 3B), suggesting that NP interacts specifically with H3. Assuming that the amount of changed fluorescence corresponds to the fraction of NP bound to H3, fitting of the binding curve (Fig. 3B) resulted in an unambiguous 1:1 stoichiometry for the interaction between H3 and HCoV-OC43 NP with a kd of 2.08 × 10−6 M. Together with the SPR results, our data indicate that H3 inhibits the RNA-binding activity of HCoV-OC43 NP by directly interacting with the NP protein.
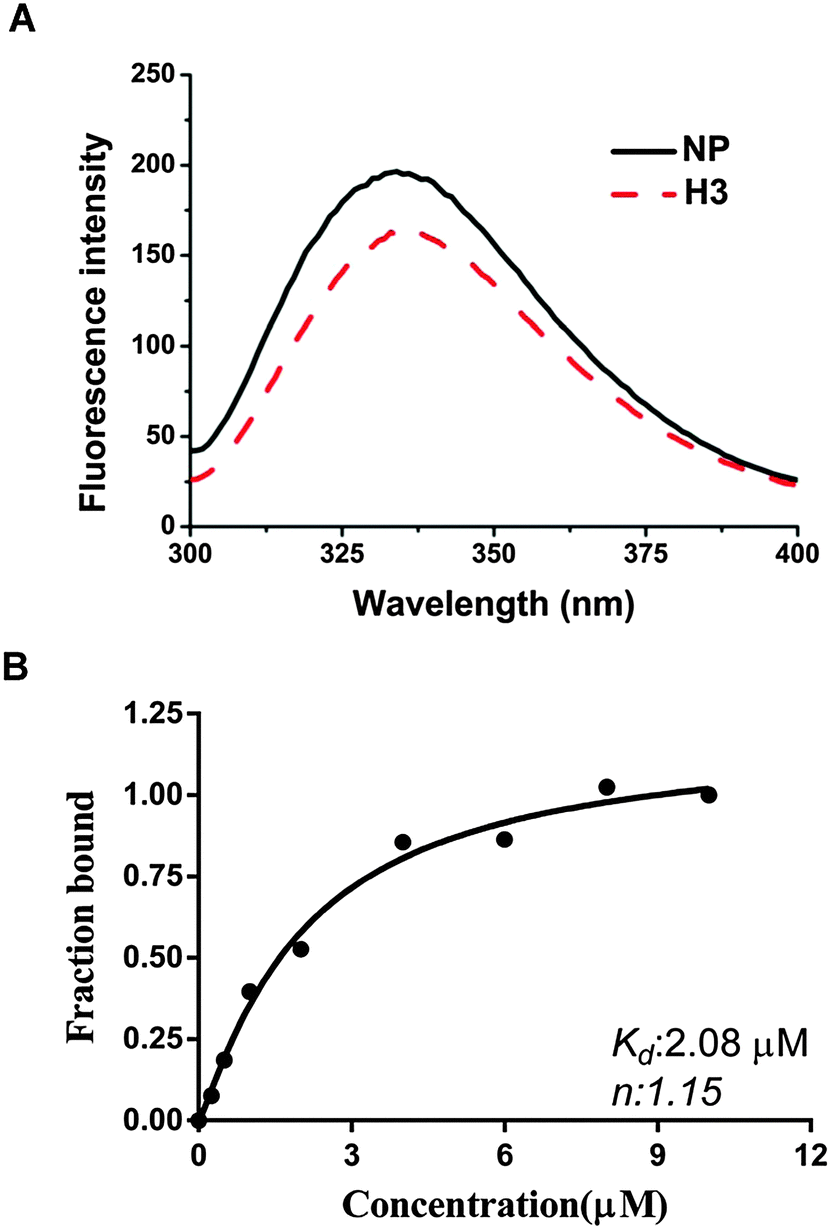 | ||
| Fig. 3 (A) FL spectra of the NP (4 μM) incubated in the presence of H3 at various concentrations for 3 h; H3 was buffered with 20 mM Tris-HCl (pH 7.5) and 100 mM NaCl. (B) Nonlinear regression analyses of the relative fluorescence intensity change of the NP in the presence of various concentrations of H3. | ||
Structure of the HCoV-OC43 NP-NTD complex with H3
To elucidate the molecular mechanism behind the binding between H3 and HCoV-OC43 NP, NP-NTD crystals were co-crystallized with H3 as described in the Materials and methods section. The structure of the HCoV-OC43 NP-NTD in complex with H3 was determined by molecular replacement using the crystal structure of the apo-HCoV-OC43 NP-NTD (PDB ID: 3V3P) as the search model. The final protein structure (Fig. 4A) was refined to R-factor and R-free values of 0.221 and 0.232, respectively, at a resolution of 2.55 Å. A five-stranded anti-parallel β sheet sandwiched between loops (or short 310 helix) forms the core of the HCoV-OC43 NP-NTD-H3 complex (Fig. 4A and B). The complex structure is shaped like a right-handed fist, in which the palm and finger regions are rich in basic residues and the β-sheet core is flanked by the flexible loops.11 Residues 105 to 120 form a long flexible loop that protrudes out of the core and connects strands β2 and β3. The loop has poor electron density probably due to its unstructured nature, making it relatively difficult to assign precise positions of the loop residues, especially for residues 115–117. The overall structure of the HCoV-OC43 NP-NTD-H3 complex is similar to that of apo-HCoV-OC43 NP-NTD (RMSD = 0.144 Å), with a H3 molecule inserted into a cavity in the nucleotide-binding site of NP-NTD (Fig. 4B).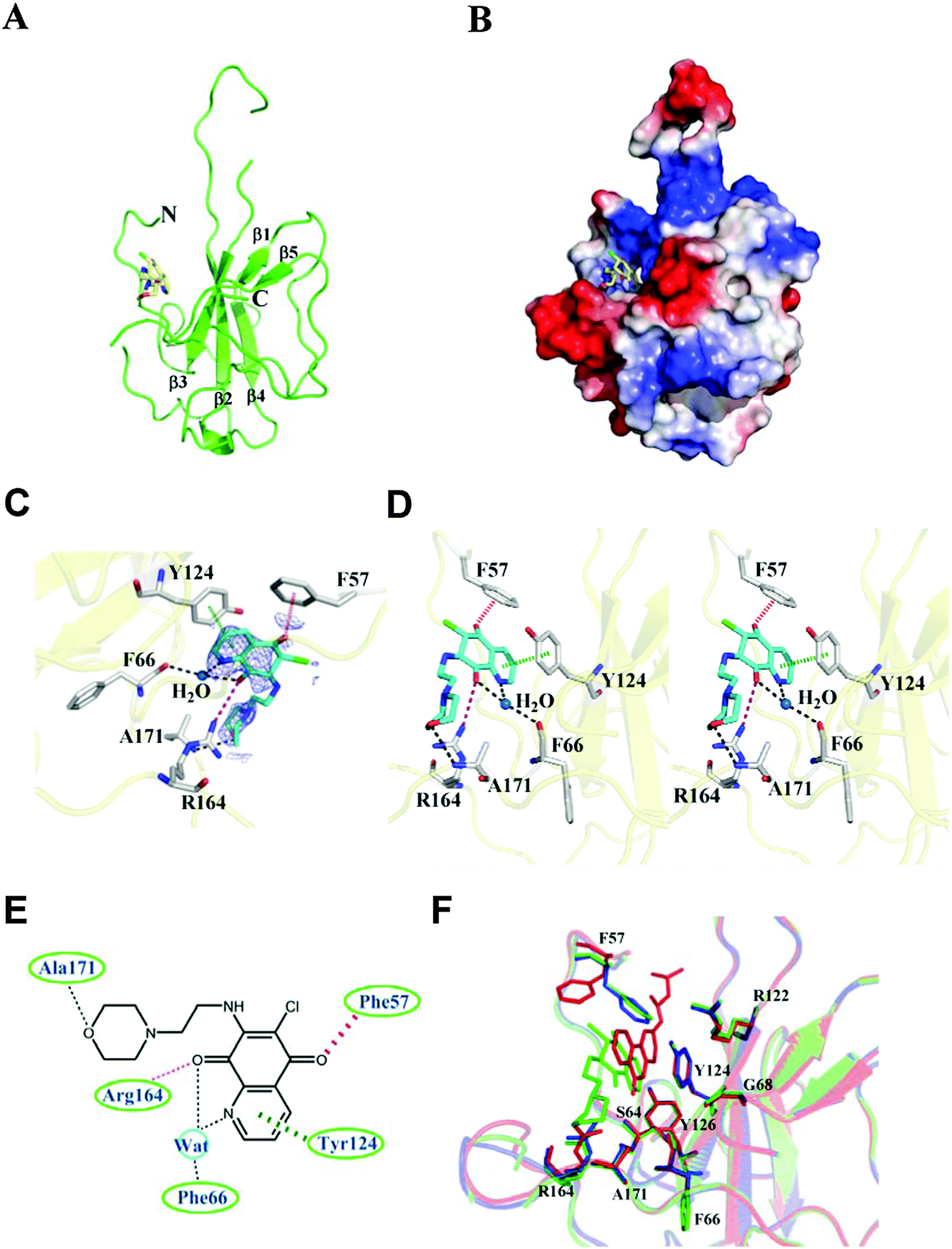 | ||
| Fig. 4 Structural overview of the HCoV-OC43 NP-NTD-H3 complex. (A) Ribbon representation of HCoV-OC43 NP-NTD with H3 depicted as a stick structure. (B) Electrostatic surface of the OC43 NP-NTD-H3 complex. Blue denotes positive charge potential, while red indicates negative charge potential. (C) 2Fo-Fc electron density map of H3 contoured at 1σ. (D) Detailed stereoview of the interactions at the H3-binding site. The H3 molecule binds to this site via Ser64, Phe66, His104, Tyr124, and Tyr126. The dotted purple and black lines denote hydrogen bonds. The red dashed lines indicate anion–π interactions. (E) Schematic diagram of H3 bound to HCoV-OC43 NP-NTD. The hydrogen-bonding interactions mediated by the side- and main-chain atoms are marked as dotted purple and black lines, respectively. The anion–π interactions mediated by the side-chain atoms are denoted as red dashed lines. The stacking interactions mediated by the side-chain atoms are marked as dashed green lines. (F) Structural superimposition of the native HCoV-OC43 NP-NTD (blue), HCoV-OC43 NP-NTD-H3 (green) and HCoV-OC43 NP-NTD-PJ34 (red) at the residues involved in ligand binding. | ||
The H3-binding pocket of NP-NTD
The H3-binding site is close to a positively charged patch which has been proposed to interact with the phosphate backbone of RNA.17 The H3-binding site has well-defined electron density (Fig. 4C), allowing a detailed analysis of the interactions between H3 and HCoV-OC43 NP-NTD (Fig. 4D). The quinoline-5,8-dione ring forms a direct hydrogen bond with the Arg164 guanidinium group and a water-mediated hydrogen bond with the backbone carbonyl group of Phe66. The oxygen atom in the H3 morpholine ring forms another hydrogen bond with the Ala171 backbone amide. Tyr124 is located on the surface of HCoV-OC43 NP-NTD and interacts directly with the quinolone group present in H3. In addition, an anion–π interaction was observed between Phe57 and the oxygen atom of the quinoxaline moiety. The side chains of Phe66, Tyr124, Arg164, Phe57 and Arg171 generate a distinct binding pocket for the interaction with H3 via hydrogen bonding, anion–π and π–π stacking forces (Fig. 4E). The specific interaction between Tyr124 and the quinolone ring of H3 was also observed in the molecular docking results, further suggesting the validity of our virtual screening approach. However, the branched moiety is pointing towards the opposite side in the docking results. Compared to a previously identified coronavirus NP inhibitor, N-(6-oxo-5,6-dihydrophenanthridin-2-yl)(N,N-dimethylamino)acetamide hydrochloride (PJ34),19 both compounds utilize a polycyclic aromatic core to interact with Phe66 and Tyr124 of NP through π–π stacking. However, the morpholin moiety of H3 and the branched moiety of PJ34 point towards opposite directions (Fig. 4F), with the orientation of H3 closely resembling that of the natural substrate AMP when bound to NP-NTD (Fig. S2, ESI†).Several studies have reported that the N-terminus of the SARS-CoV NP provides a scaffold for RNA binding.25,33 X-ray crystallographic studies have also revealed a high degree of conservation among structural folds of NP-NTDs from various CoVs. The N-terminal region of IBV NP assists in the formation of the ribonucleocapsid via a lure and lock mechanism by facilitating long-range, non-specific interactions between the NP and viral RNA.19 Multiple RNA binding sites on the NP of SARS-CoV have been identified that may bind to RNA cooperatively.34 Similar to the N-terminal domain of SARS-CoV and IBV NPs, the N-terminal domain of the HCoV-OC43 NP may also bind to RNA. In addition to the N-terminal domain, the highly positively charged central linker region was also capable of binding to RNA, whereas the C-terminal domain of the HCoV-OC43 NP did not show RNA-binding activity.34 As such, identification of compounds that bind to the N-terminal domain and interfere with NP–RNA interactions may provide valuable leads into development of broad-spectrum anti-coronaviral therapeutics. Several residues in NP-NTDs are critical for RNA binding and virus infectivity. We have previously shown that compounds binding to a unique RNA-binding pocket located in the center of CoV NP-NTD may be employed to combat pathogenic CoVs.28
In this study, docking results suggest that several hits can bind to this ribonucleotide-binding pocket of CoV NP-NTD. Based on the SPR assay of NPs, we identified one potential compound that interferes with the RNA binding activity of CoV NP.28 Previously, we identified another CoV NP inhibitor, PJ34, using virtual screening.28 The higher resemblance between H3 and AMP compared to that between PJ34 and AMP may explain the higher inhibitory effect of H3 towards the RNA-binding activity of HCoV-OC43 NP at the same protein-to-compound ratios (Fig. S2, ESI†). Interestingly, PJ34 (compound H5) induced less fluorescence quenching compared to H3 (Fig. 1B), which suggests that the extent of change induced by compounds may correlate with their relative inhibitory activity.
Conclusion
In summary, we identified a new NP inhibitor obtained from in silico virtual screening against CoV NPs and the results were further verified experimentally with the SPR assay and X-ray crystallography. We also characterized the binding affinity of the compound with CoV NP by fluorescence studies suggesting small compound H3 as an effective inhibitor for the RNA-binding activity of CoV NPs. The development of novel NP-targeting agents is crucial for treating pathogenic HCoV infections and persistence of latent infection – problems that have been highlighted in the field of influenza and AIDS research. This study may assist in the development of anti-viral drugs against HCoV. Based on this aspect, our findings will help explore the role of CoV NPs in ribonucleoprotein formation, metabolism and its multi-functional role in the eukaryotic system.Conflict of interest
None.Acknowledgements
This work was supported by NSC grant (103-2113-M-005-007-MY3) (M.-H. H). We would like to thank the National Synchrotron Radiation Research Center (Taiwan) for X-ray data collection.References
- F. W. Denny and F. A. Loda, Am. J. Trop. Med. Hyg., 1986, 35, 1–2 CAS.
- B. G. Williams, E. Gouws, C. Boschi-Pinto, J. Bryce and C. Dye, Lancet Infect. Dis., 2002, 2, 25–32 CrossRef PubMed.
- B. D. Gessner, Expert Rev. Respir. Med., 2011, 5, 459–463 CrossRef PubMed.
- K. E. Arden, P. McErlean, M. D. Nissen, T. P. Sloots and I. M. Mackay, J. Med. Virol., 2006, 78, 1232–1240 CrossRef CAS PubMed.
- M. Owusu, A. Annan, V. M. Corman, R. Larbi, P. Anti, J. F. Drexler, O. Agbenyega, Y. Adu-Sarkodie and C. Drosten, PLoS One, 2014, 9, e99782 Search PubMed.
- M. Venter, R. Lassauniere, T. L. Kresfelder, Y. Westerberg and A. Visser, J. Med. Virol., 2011, 83, 1458–1468 CrossRef PubMed.
- H. Smuts, Influenza Other Respir. Viruses, 2008, 2, 135–138 CrossRef PubMed.
- H. Smuts, L. Workman and H. J. Zar, J. Med. Virol., 2008, 80, 906–912 CrossRef CAS PubMed.
- M. J. Adams and E. B. Carstens, Arch. Virol., 2012, 157, 1411–1422 CrossRef CAS PubMed.
- A. Zumla, D. S. Hui and S. Perlman, Lancet, 2015, 386, 995–1007 CrossRef.
- C. M. Coleman and M. B. Frieman, J. Virol., 2014, 88, 5209–5212 CrossRef PubMed.
- Z. Lou, Y. Sun and Z. Rao, Trends Pharmacol. Sci., 2014, 35, 86–102 CrossRef CAS PubMed.
- S. Milne-Price, K. L. Miazgowicz and V. J. Munster, Pathog. Dis., 2014, 71, 121–136 CrossRef PubMed.
- P. S. Masters, M. M. Parker, C. S. Ricard, C. Duchala, M. F. Frana, K. V. Holmes and L. S. Sturman, Adv. Exp. Med. Biol., 1990, 276, 239–246 CrossRef CAS PubMed.
- Z. Hayouka, J. Rosenbluh, A. Levin, S. Loya, M. Lebendiker, D. Veprintsev, M. Kotler, A. Hizi, A. Loyter and A. Friedler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8316–8321 CrossRef CAS PubMed.
- C. K. Chang, M. H. Hou, C. F. Chang, C. D. Hsiao and T. H. Huang, Antiviral Res., 2014, 103, 39–50 CrossRef CAS PubMed.
- I. J. Chen, J. M. Yuann, Y. M. Chang, S. Y. Lin, J. Zhao, S. Perlman, Y. Y. Shen, T. H. Huang and M. H. Hou, Biochim. Biophys. Acta, 2013, 1834, 1054–1062 CrossRef CAS PubMed.
- Y. Ma, X. Tong, X. Xu, X. Li, Z. Lou and Z. Rao, Protein Cell, 2010, 1, 688–697 CrossRef CAS PubMed.
- K. S. Saikatendu, J. S. Joseph, V. Subramanian, B. W. Neuman, M. J. Buchmeier, R. C. Stevens and P. Kuhn, J. Virol., 2007, 81, 3913–3921 CrossRef CAS PubMed.
- H. Jayaram, H. Fan, B. R. Bowman, A. Ooi, J. Jayaram, E. W. Collisson, J. Lescar and B. V. Prasad, J. Virol., 2006, 80, 6612–6620 CrossRef CAS PubMed.
- I. M. Yu, M. L. Oldham, J. Zhang and J. Chen, J. Biol. Chem., 2006, 281, 17134–17139 CrossRef CAS PubMed.
- H. Fan, A. Ooi, Y. W. Tan, S. Wang, S. Fang, D. X. Liu and J. Lescar, Structure, 2005, 13, 1859–1868 CrossRef CAS PubMed.
- Y. S. Wang, C. K. Chang and M. H. Hou, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 977–980 CAS.
- N. E. Grossoehme, L. Li, S. C. Keane, P. Liu, C. E. Dann 3rd, J. L. Leibowitz and D. P. Giedroc, J. Mol. Biol., 2009, 394, 544–557 CrossRef CAS PubMed.
- Q. Huang, L. Yu, A. M. Petros, A. Gunasekera, Z. Liu, N. Xu, P. Hajduk, J. Mack, S. W. Fesik and E. T. Olejniczak, Biochemistry, 2004, 43, 6059–6063 CrossRef CAS PubMed.
- S. Chenavas, T. Crepin, B. Delmas, R. W. Ruigrok and A. Slama-Schwok, Future Microbiol., 2013, 8, 1537–1545 CrossRef CAS PubMed.
- Y. S. Lo, S. Y. Lin, S. M. Wang, C. T. Wang, Y. L. Chiu, T. H. Huang and M. H. Hou, FEBS Lett., 2013, 587, 120–127 CrossRef CAS PubMed.
- S. Y. Lin, C. L. Liu, Y. M. Chang, J. Zhao, S. Perlman and M. H. Hou, J. Med. Chem., 2014, 57, 2247–2257 CrossRef CAS PubMed.
- L. Pu, A. A. Amoscato, M. E. Bier and J. S. Lazo, J. Biol. Chem., 2002, 277, 46877–46885 CrossRef CAS PubMed.
- J. S. Lazo, D. C. Aslan, E. C. Southwick, K. A. Cooley, A. P. Ducruet, B. Joo, A. Vogt and P. Wipf, J. Med. Chem., 2001, 44, 4042–4049 CrossRef CAS PubMed.
- Y. S. Lo, W. H. Tseng, C. Y. Chuang and M. H. Hou, Nucleic Acids Res., 2013, 41, 4284–4294 CrossRef CAS PubMed.
- H. C. Hung, C. L. Liu, J. T. Hsu, J. T. Horng, M. Y. Fang, S. Y. Wu, S. H. Ueng, M. Y. Wang, C. W. Yaw and M. H. Hou, Anal. Chem., 2012, 84, 6391–6399 CrossRef CAS PubMed.
- C. K. Chang, Y. L. Hsu, Y. H. Chang, F. A. Chao, M. C. Wu, Y. S. Huang, C. K. Hu and T. H. Huang, J. Virol., 2009, 83, 2255–2264 CrossRef CAS PubMed.
- C. Y. Huang, Y. L. Hsu, W. L. Chiang and M. H. Hou, Protein Science: A Publication of the Protein Society, 2009, 18, 2209–2218 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The atomic coordinates and structure factors for OC43 complexes with H3 (4LMT) have been deposited in the RCSB Protein Data Bank. See DOI: 10.1039/c5mb00582e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |