
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of the ionic liquid [BMP][TFSA] with rutile TiO2(110) and coadsorbed lithium†
Benedikt
Uhl
ab,
Maral
Hekmatfar
ab,
Florian
Buchner
ab and
R. Jürgen
Behm
*ab
aInstitute of Surface Chemistry and Catalysis, Ulm University, Albert-Einstein-Allee 47, D-89081 Ulm, Germany. E-mail: juergen.behm@uni-ulm.de; Fax: +49-731-50-25452
bHelmholtz-Institute Ulm (HIU) Electrochemical Energy Storage, Helmholtzstr. 11, D-89081 Ulm, Germany
First published on 28th January 2016
Abstract
Aiming at a fundamental understanding of the processes at the electrode|ionic liquid interface in Li ion batteries, we investigated the interaction of the ionic liquid n-butyl-n-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide [BMP][TFSA] and of Li with a reduced rutile TiO2(110) (1 × 1) surface as well as the interaction between [BMP][TFSA] and Li on the TiO2(110) surface under ultrahigh vacuum (UHV) conditions by X-ray photoelectron spectroscopy and scanning tunnelling microscopy. Between 80 K and 340 K [BMP][TFSA] adsorbs molecularly on the surface and at higher temperatures decomposition is observed, resulting in products such as Sad, Fad and TiNx. The decomposition pattern is compared to proposals based on theory. Small amounts of Li intercalate even at 80 K into TiO2(110), forming Li+ and Ti3+ species. The stoichiometry in the near surface region corresponds to Li7Ti5O12. For higher coverages in the range of several monolayers part of the Li remains on the surface, forming a Li2O cover layer. At 300 K, Ti3+ species become sufficiently mobile to diffuse into the bulk. Li post-deposition on a [BMP][TFSA] covered TiO2(110) surface at 80 K results in two competing reactions, Li intercalation and reaction with the IL, resulting in the decomposition of the IL. Upon warming up, the Ti3+ formed at low T is consumed by reaction with the IL adlayer and intermediate decomposition products. Post-deposition of [BMP][TFSA] (300 K) on a surface pre-covered with a Li2O/Li7Ti5O12 layer results in the partial reaction of [BMP][TFSA] with the Li+ and Ti3+ species, which gets completed at higher temperatures.
1 Introduction
Ionic liquids, which are molten salts with a melting point below 100 °C,1 have attracted high interest in the last few decades because of their uncommon physicochemical properties such as their low vapour pressure, their non-flammability or their high electrochemical stability, and the ability to fine tune these properties by combining different anion–cation pairs.2–4 Because of the latter properties they also emerged as promising new solvents for use as electrolytes in lithium ion and lithium air batteries.5,6 In lithium ion batteries the interface between the electrolyte and the electrode and the formation of the so-called solid-electrolyte interphase (SEI), which is a protective layer of decomposition products formed in the first cycle of operation and which prevents further decomposition of the electrolyte, are crucial for the performance and stability of the battery. Despite significant efforts, a molecular scale understanding of structure, composition and formation of these protective layers is still lacking.7,8 Aiming at a comprehensive understanding of the processes directly at the interface between the solid electrode and the ionic liquid based electrolyte, we have therefore started to investigate the interactions between solids and ionic liquids on a fundamental scale, using well defined model systems and conditions. In a first step we have investigated the interaction of battery relevant ionic liquids such as n-butyl-n-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide [BMP][TFSA], 1-ethyl-3-methylimidazolium bis(trifluoro-methylsulfonyl)imide [EMIM][TFSA] and 1-octyl-3-methylimidazolium bis(trifluoromethyl-sulfonyl)imide [OMIM][TFSA] and Ag(111), Au(111) and Cu(111) model substrates under ultrahigh vacuum (UHV) conditions.9–13 Employing Surface Science tools such as scanning tunnelling microscopy (STM), angle resolved X-ray photoelectron spectroscopy (XPS) and IR spectroscopy, and combining the experimental efforts with theory we could gain detailed insight into the molecule–molecule and molecule–substrate interactions and the structure formation and thermal stability of the IL adlayers on these simplified model systems. We now extended this study to more realistic materials. In the present contribution we report the results of a study on the interaction of [BMP][TFSA] with a single crystalline rutile TiO2(110) substrate, and the effect of Li on the IL–TiO2(110) adsorption system. TiO2 with rutile or anatase structure is in addition to the spinel Li4Ti5O12 (LTO) a standard electrode material for Li ion batteries.14–16Despite extensive literature on the physics and chemistry of TiO2 surfaces,17,18 very little is known on the interaction of Li with these surfaces. Therefore, in addition to investigating the interaction between [BMP][TFSA] and TiO2(110) (Section 2.1), we also explored the interaction between Li and that surface, upon vapor deposition of small amounts of Li on that surface and upon subsequent annealing (Section 2.2). Finally, in order to gain information on the effect of Li on the interaction between a [BMP][TFSA] adlayer and TiO2(110), we investigated the interaction of [BMP][TFSA] with a TiO2(110) surface modified by pre-deposited Li, and the effect of post-deposited Li on a [BMP][TFSA] adlayer covered TiO2(110) substrate, and the thermal stability of these systems (Section 2.3).
Before presenting the present results, we will briefly summarize the main previous findings relevant for this study. The structure of ILs in the bulk and also at interfaces have been reviewed recently by Hayes et al.19 UHV studies on the interaction of Li with TiO2(110) upon Li deposition reported partly contradictory results. In a combined metastable impact electron spectroscopy (MIES) and ultraviolet photoelectron spectroscopy (UPS) study Krischok et al. reported that Li adsorbs ionically up to 0.3 ML and 130 K, accompanied by the formation of Ti3+ species at the surface. Above 160 K the Li2s signal in the MIES spectra was found to vanish, pointing to Li insertion into the substrate.20 In contrast, employing STM, Tatsumi et al. reported the observation of small protrusions on a TiO2(110) (1 × 1) surface upon Li adsorption at room temperature, which they attributed to adsorbed Li.21 In an electrochemical environment, Li intercalation was found to take place in TiO2 substrates (rutile as well as anatase) at room temperature.22–24 For rutile this intercalation was limited by slow diffusion of the Li into the bulk crystal.25,26 Based on these findings, lithium intercalation could therefore be a competing process in addition to the expected reaction with the adsorbed IL layer. Former studies of [OMIM][TFSA] in contact with bulk lithium27 and of co-deposited Li and a [BMP][TFSA] adlayer on Cu(111)28 revealed a decomposition reaction of the respective IL adlayer on the Li substrate or induced by coadsorbed Li, and the formation of various decomposition products such as LiF, Fad, Li2S, Sad, SOx,ad, CF3,ad, Li2O and LixCHy.
2 Results
2.1 [BMP][TFSA] on rutile TiO2(110)
First we focus on the interaction of [BMP][TFSA] with the rutile TiO2(110) surface. In Fig. 1 the spectra represent the XPS signals in the O1s, C1s, N1s, S2p and F1s regions, recorded at room temperature on a TiO2(110) surface covered with ∼1.5 monolayers (ML) of [BMP][TFSA], where 1 ML corresponds to a closed layer of adsorbed [BMP][TFSA]. Additionally we show a ball and stick model of a [BMP][TFSA] ion pair. The O1s region reveals two peaks at binding energies (BEs) of 530.6 eV and 532.8 eV. The peak at lower BE is related to the oxide ions of the TiO2 substrate. Its position agrees with values reported for a clean and fully oxidized rutile TiO2 crystal (530.4 eV).29 The peak at 532.8 eV as well as the peaks in the other regions shown in Fig. 1 appear only after evaporation of [BMP][TFSA] on the substrate. The peaks resemble what is expected for adsorption of the intact [BMP][TFSA] molecules: the peak at 532.8 eV in the O1s region is attributed to the SO2 groups of the [TFSA]− anion. In the C1s region three peaks are identified, similar to previous reports for [BMP][TFSA] multilayer adsorption on Cu(111)12 and Ag(111):10 the peaks at Eb = 285.6 eV and 287 eV arise from the [BMP]+ cations, where the C-atoms directly bonding to the N-atom (“Chetero”) have a 1.4 eV higher BE than the alkyl C-atoms (“Calkyl”). The peak at 293.2 eV is related to the C-atoms in the [TFSA]− anion. As expected for the CF3 groups with their highly electronegative substituents, this peak is strongly shifted to higher BE compared to the cation related peaks. In the N1s region two peaks with equal intensity appear at 402.8 eV and 399.7 eV. Since the electron density at the positively charged N-atom in the cation is lower than at the N-atom in the anion, the peak with higher BE is attributed to Ncation, the other one to Nanion. This assignment resembles earlier ones reported for [BMP][TFSA] on Cu(111)28 and for [xMIM][TFSA] (xMIM = 1x-3methylimidazolium with x = methyl, ethyl, octyl) on Au(111), Ni(111) and a glass substrate, respectively.30–32 A doublet peak in the S2p region, at 169.3 eV (S2p3/2) and 170.5 eV (S2p1/2), and a singlet peak in the F1s region, at 689.1 eV, originate from the [TFSA]− anion. After correcting for the different cross sections/sensitivity factors of the different peaks, their intensities largely fit (±0.2) to the stoichiometry of the intact [BMP][TFSA] ion pair, with a composition of 4 OTFSA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Canion:4 Chetero:5 Calkyl:1 Ncation:1 Nanion:2 STFSA:6 FTFSA. Only for FTFSA, we find a higher intensity (6.6–7) than expected from the stoichiometry, while for OTFSA the relative intensity (3.2) is lower than expected. Similar XPS results, including the deviations from a stoichiometric intensity distribution, were reported and discussed by Cremer et al. for [OMIM][TFSA] and [MMIM][TFSA] on Au(111)31 and Ni(111).32 Following their interpretation these deviations can be used to draw conclusions on the adsorption geometry of the [BMP][TFSA] adsorbates. Elements with signal intensities higher than expected from the stoichiometry are in average positioned slightly higher in the adsorbate layer, those with lower signal intensities closer to the surface with their XPS signal damped by inelastic scattering at the atoms located higher. Accordingly, we assume an adsorption geometry similar to those found for [MMIM][TFSA] on Au(111)31 and for [BMP][TFSA] on Au(111)9 and Ag(111)10 with both the anion and the cation directly adsorbed on the surface. Following our earlier discussion for adsorption on Ag(111),10 the anion adopts a cis-conformation (as shown in Fig. 1), with the O atoms pointing to the surface, and the F atoms to the vacuum. The other atoms are arranged on the main axis of the molecule and are roughly positioned at the same height relative to the substrate surface. In this geometry, the signal of the O atoms is damped relative to the signals of the other atoms, while that of the F atoms is more intense. (In this discussion it should be kept in mind that the XPS data were recorded at a very shallow emission angle of 80° to the surface normal, which makes the measurement very surface sensitive.) Since the intensities of the cation related peaks also fit to the stoichiometry, the cation atoms should be arranged roughly at the same height above the surface as the atoms in the main axis of the anion. For the vertical position of the Calkyl atoms, which are situated in the butyl chain and in the ring of the cation, we can only state that in average they are at the same height as the Chetero atoms. The individual positions of the different Calkyl atoms may be different, if, e.g., a higher position of the atoms in the alkyl chain is counterbalanced by a lower position of the Calkyl atoms in the ring. Note that this means that from the XPS measurements we cannot decide if the alkyl chain is adsorbed flat on the surface or if it is pointing up from the surface.
:2 Canion:4 Chetero:5 Calkyl:1 Ncation:1 Nanion:2 STFSA:6 FTFSA. Only for FTFSA, we find a higher intensity (6.6–7) than expected from the stoichiometry, while for OTFSA the relative intensity (3.2) is lower than expected. Similar XPS results, including the deviations from a stoichiometric intensity distribution, were reported and discussed by Cremer et al. for [OMIM][TFSA] and [MMIM][TFSA] on Au(111)31 and Ni(111).32 Following their interpretation these deviations can be used to draw conclusions on the adsorption geometry of the [BMP][TFSA] adsorbates. Elements with signal intensities higher than expected from the stoichiometry are in average positioned slightly higher in the adsorbate layer, those with lower signal intensities closer to the surface with their XPS signal damped by inelastic scattering at the atoms located higher. Accordingly, we assume an adsorption geometry similar to those found for [MMIM][TFSA] on Au(111)31 and for [BMP][TFSA] on Au(111)9 and Ag(111)10 with both the anion and the cation directly adsorbed on the surface. Following our earlier discussion for adsorption on Ag(111),10 the anion adopts a cis-conformation (as shown in Fig. 1), with the O atoms pointing to the surface, and the F atoms to the vacuum. The other atoms are arranged on the main axis of the molecule and are roughly positioned at the same height relative to the substrate surface. In this geometry, the signal of the O atoms is damped relative to the signals of the other atoms, while that of the F atoms is more intense. (In this discussion it should be kept in mind that the XPS data were recorded at a very shallow emission angle of 80° to the surface normal, which makes the measurement very surface sensitive.) Since the intensities of the cation related peaks also fit to the stoichiometry, the cation atoms should be arranged roughly at the same height above the surface as the atoms in the main axis of the anion. For the vertical position of the Calkyl atoms, which are situated in the butyl chain and in the ring of the cation, we can only state that in average they are at the same height as the Chetero atoms. The individual positions of the different Calkyl atoms may be different, if, e.g., a higher position of the atoms in the alkyl chain is counterbalanced by a lower position of the Calkyl atoms in the ring. Note that this means that from the XPS measurements we cannot decide if the alkyl chain is adsorbed flat on the surface or if it is pointing up from the surface.
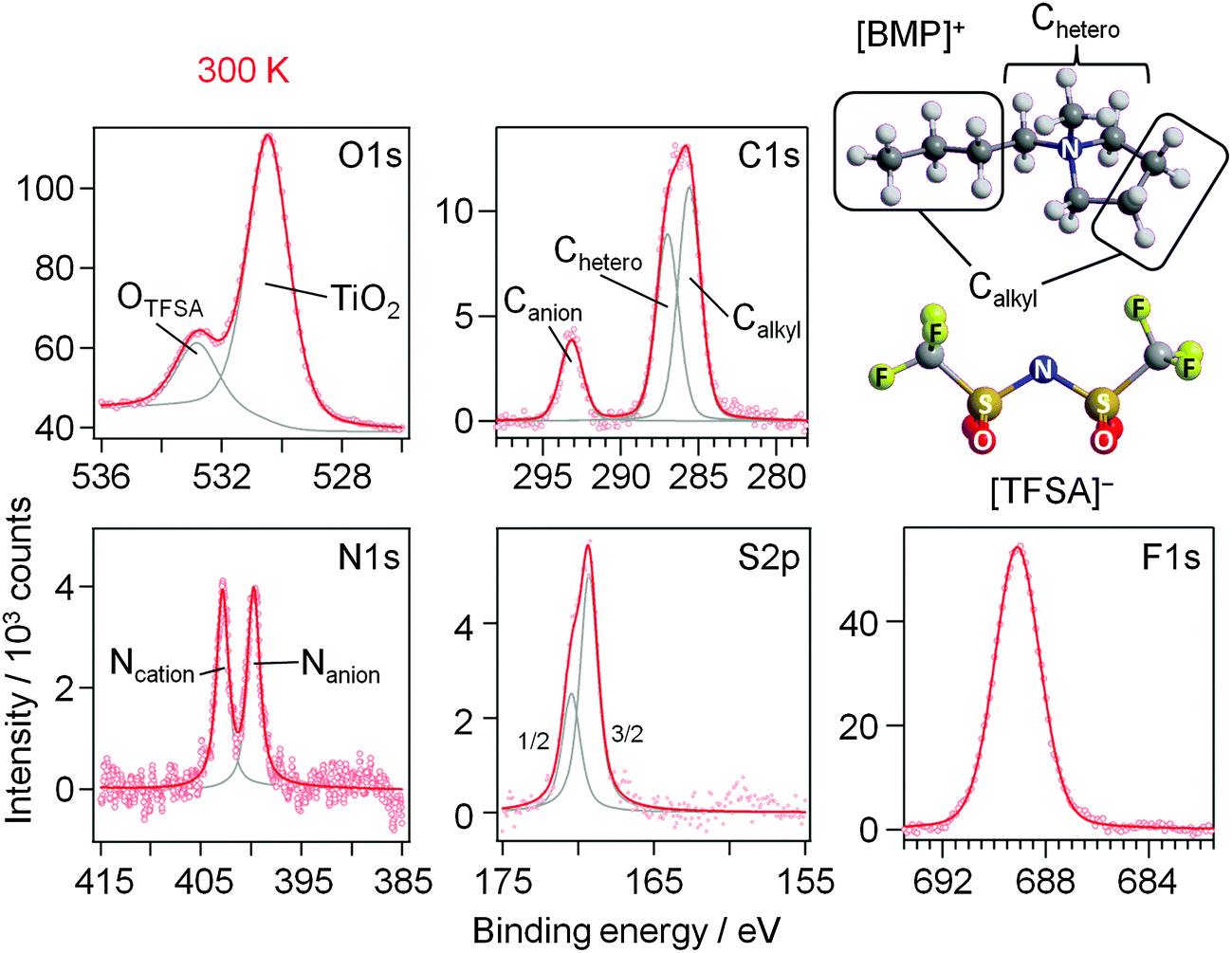 | ||
| Fig. 1 O1s, C1s, N1s, S2p and F1s XP core level spectra of 1.5 ML [BMP][TFSA] adsorbed on TiO2(110) at 300 K. A structural model of [BMP][TFSA] is displayed on the right hand side. | ||
Additional information on the adsorption behaviour of [BMP][TFSA] on rutile(110) was obtained from STM measurements. STM images of a clean rutile(110) surface, which illustrate also the quality of the surface preparation, are shown in Fig. 2a and b. In Fig. 2a the STM contrast resembles that known from various publications.17,18,33 There is a consensus that the line structure observed in the STM images is due to a (1 × 1) structure. A detailed model was given in ref. 18. In short, the terminating atoms along the [001] direction are aligned in rows of (a) twofold coordinated “bridge-bonded” O-atoms on top of 6-fold coordinated Ti-atoms, (b) 3-fold coordinated O-atoms and (c) 5-fold coordinated Ti-atoms in the sequence …abcbabcb… along the [1−10] direction. The bridge-bonded oxygen rows are geometrically highest, but appear as dark rows in STM images, while the (lower lying) 5-fold coordinated Ti-atoms are represented by the rows of bright protrusions. The appearance of the STM images therefore is opposite to the topography of the surface, meaning that it is dominated by electronic effects. The individual bright spots, which are always positioned on top of the dark rows, have been explained as missing bridge-bonded oxygen atoms at the surface (oxygen vacancies).17
 | ||
| Fig. 2 (a) STM image of a clean TiO2(110) surface. The typical lines of the (1 × 1) structure are clearly visible as well as bright spots, indicating oxygen vacancies in the bridge-bonded oxygen rows (T = 110 K, UT = 1.1 V, IT = 21 pA). (b) Inverted contrast STM image of TiO2(110) with atomic resolution of the bridge-bonded oxygen atoms and dark spots in these rows resembling missing bridge-bonded oxygen atoms (T = 300 K, UT = 1.7 V, IT = 650 pA). Small (c) and large scale (d) STM images of TiO2(110) covered with 1 ML of [BMP][TFSA] (c: T = 127 K, UT = 1.1 V, IT = 20 pA; d: T = 126 K, UT = 1.1 V, IT = 20 pA). | ||
In addition to the STM images with “normal” contrast (see above), we sometimes also obtained images as shown in Fig. 2b, where the rows of bridge-bonded oxygen atoms appear as bright lines. Images with comparable contrast had also been reported previously;33,34 by some authors they were termed as STM images with ‘non-reversed contrast’.34 These images closely resembled images of the TiO2(110) surface obtained by non-contact atomic force microscopy (NC-AFM) with atomical resolution.35 Since the tunnelling parameters are in general not different from those used in the “normal” reversed-contrast images, this different contrast is presumably due to changing tip conditions. Overall, the STM images largely resemble those reported in the literature for (1 × 1) surface structure on a partly reduced sample.36–40
Room temperature STM imaging of an [BMP][TFSA] adlayer covered TiO2(110) surface, which had been prepared by [BMP][TFSA] evaporation at room temperature, results only in noisy features on top of the (1 × 1) surface structure. We explain this by the formation of a 2D gas/liquid on the surface, similar to previous findings for [BMP][TFSA], [EMIM][TFSA] and [OMIM][TFSA] on Au(111) and Ag(111).9,10,13 In the latter cases, cooling down to (nearly) liquid nitrogen (LN2) temperatures led to the formation of condensed 2D crystalline and 2D glass phases or islands on the surface. For [BMP][TFSA] on TiO2(110), even STM imaging at 100 K resulted in very noisy STM images. Since we were reproducibly able to clearly resolve the clean TiO2(110) surface, we do not think that this is a technical problem of the STM measurement, but indicates that even at this low temperature the adlayer is rather mobile, forming a mobile 2D gas/liquid phase. In that case, the mobility of [BMP][TFSA] is significantly higher on the TiO2(110) surface than on the noble metal surfaces investigated so far.9,10 Only at coverages close to 1 ML we could resolve new features, and only at very low tunnelling currents <20 pA, as shown in Fig. 2c. Most likely, the formation of reasonably stable structures is related to a stabilization of the adsorbates in the closed layers by adsorbate–adsorbate interactions. In the STM image round protrusions can be resolved, which are essentially closely packed along the [001] oriented rows formed by the (1 × 1) surface structure of TiO2(110). The distance in the [1−10] direction between the rows was 0.61 ± 0.04 nm. This fits very well to the distance between the bridge-bonded oxygen rows of the TiO2(110) surface of 0.63 ± 0.02 nm (literature: 0.63 ± 0.025 nm18). Along the rows the distances seem to vary at around 0.61 ± 0.07 nm. The diameter and density of the round protrusions are measured from several independent STM images to be 0.56 ± 0.06 nm (full width at half maximum) and 2.6 ± 0.4 nm−2. The round protrusions observed in these images shall be compared with the structures formed for [BMP][TFSA] on Au(111) and Ag(111),11 where in addition to round protrusions also pairs of longish protrusions appeared next to each other on the surface at LN2 temperature. Based also on DFT-D calculations,10 the round protrusions were identified as [BMP]+ cations adsorbed on Ag(111), while a pair of longish protrusions reflects a [TFSA]− anion. The density of cations was 0.61–0.79 nm−2 (depending on the substrate and the coverage), and their diameter in the STM images (full width at half maximum) was 0.70 ± 0.04 nm. Our observations shall further be compared with the results of an in situ electrochemical study of [BMP][TFSA] adsorption on Au(111), where two different adlayer structures were found: at the negatively charged interface at −1.4 V vs. a Pt quasi reference, only the [BMP]+ cations adsorb in the first layer in a 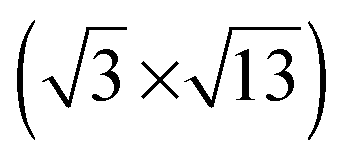 superstructure with a density of 1.9 nm−2 cations, with the alkyl chains lying flat on the surface. For adsorption at −1.6 V they reported a
superstructure with a density of 1.9 nm−2 cations, with the alkyl chains lying flat on the surface. For adsorption at −1.6 V they reported a 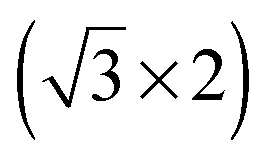 structure with a density of 3.6 cations nm−2, with the alkyl chains pointing upwards from the surface.41 Also monolayers of a related IL, [BMP][FAP] ([BMP]+-tris(pentafluoroethyl)trifluorophosphate), on Au(111)42 (same cation, different anion) appeared in STM images as randomly distributed round protrusions with a diameter of ∼0.5 nm, a mean distance of 0.45 ± 0.02 nm between the protrusions and a density of 4.5 ± 0.1 nm−2 (imaged under UHV conditions at 210 K). There it was discussed whether the protrusions represent both ion types (1 protrusion = 1 [BMP]+ or 1 [FAP]−) or whether the ions form two layers on top of each other, where STM detects only the upper layer. The possibility that one protrusion represents a complete ion pair was ruled out because the size of the protrusion and their mean distance are too small. The density of protrusions found for [BMP][TFSA] on TiO2(110) is between that of the adsorbed cations on the charged Au(111) surfaces, where it is higher (138% of that on TiO2(110)) at −1.6 V and lower (73%) at −1.4 V. On the other hand, it is much higher (373%) than in the structures of [BMP][TFSA] on Au(111) and Ag(111) at the solid|UHV interface with both the anion and cation directly adsorbed on the surface. It should also be noted that the round protrusions have roughly the same size as the ones representing [BMP]+ cations adsorbed with upstanding alkyl chains on Au(111) and Ag(111). Considering that based on the XPS results both cations and anions are directly adsorbed on the TiO2(110) surface, the present STM results seem to indicate that on TiO2(110) one [BMP][TFSA] ion pair is represented by either 3 or 4 round protrusions. In both cases we assume that the anion appears as two protrusions, close to previous findings on Au(111) and Ag(111), where the anion is represented by two protrusions with longish shapes and a lower height compared to the round protrusions representing the cation alkyl chain.9,10 In the first case the remaining one of the three round protrusions per ion pair would represent an adsorbed [BMP]+ cation with the alkyl chain pointing upwards. The second case could be rationalized by a model where the cation is imaged as two protrusions, one for the ring and one for the alkyl chain, in this case most likely a flat lying alkyl chain. That would lead to densities of 0.87 or 0.65 ion pairs nm−2, respectively, which is in the range of the densities of adsorbed ion pairs with upstanding alkyl chain on the Au(111) and Ag(111) surfaces under UHV conditions. Because of the much higher noise level in these images compared to those obtained for [BMP][TFSA] on Au(111) and Ag(111), a final decision on the adlayer structure on the molecular level is not possible at present.
structure with a density of 3.6 cations nm−2, with the alkyl chains pointing upwards from the surface.41 Also monolayers of a related IL, [BMP][FAP] ([BMP]+-tris(pentafluoroethyl)trifluorophosphate), on Au(111)42 (same cation, different anion) appeared in STM images as randomly distributed round protrusions with a diameter of ∼0.5 nm, a mean distance of 0.45 ± 0.02 nm between the protrusions and a density of 4.5 ± 0.1 nm−2 (imaged under UHV conditions at 210 K). There it was discussed whether the protrusions represent both ion types (1 protrusion = 1 [BMP]+ or 1 [FAP]−) or whether the ions form two layers on top of each other, where STM detects only the upper layer. The possibility that one protrusion represents a complete ion pair was ruled out because the size of the protrusion and their mean distance are too small. The density of protrusions found for [BMP][TFSA] on TiO2(110) is between that of the adsorbed cations on the charged Au(111) surfaces, where it is higher (138% of that on TiO2(110)) at −1.6 V and lower (73%) at −1.4 V. On the other hand, it is much higher (373%) than in the structures of [BMP][TFSA] on Au(111) and Ag(111) at the solid|UHV interface with both the anion and cation directly adsorbed on the surface. It should also be noted that the round protrusions have roughly the same size as the ones representing [BMP]+ cations adsorbed with upstanding alkyl chains on Au(111) and Ag(111). Considering that based on the XPS results both cations and anions are directly adsorbed on the TiO2(110) surface, the present STM results seem to indicate that on TiO2(110) one [BMP][TFSA] ion pair is represented by either 3 or 4 round protrusions. In both cases we assume that the anion appears as two protrusions, close to previous findings on Au(111) and Ag(111), where the anion is represented by two protrusions with longish shapes and a lower height compared to the round protrusions representing the cation alkyl chain.9,10 In the first case the remaining one of the three round protrusions per ion pair would represent an adsorbed [BMP]+ cation with the alkyl chain pointing upwards. The second case could be rationalized by a model where the cation is imaged as two protrusions, one for the ring and one for the alkyl chain, in this case most likely a flat lying alkyl chain. That would lead to densities of 0.87 or 0.65 ion pairs nm−2, respectively, which is in the range of the densities of adsorbed ion pairs with upstanding alkyl chain on the Au(111) and Ag(111) surfaces under UHV conditions. Because of the much higher noise level in these images compared to those obtained for [BMP][TFSA] on Au(111) and Ag(111), a final decision on the adlayer structure on the molecular level is not possible at present.
The 2D growth behaviour of [BMP][TFSA] adsorbates at low temperatures was also examined in large-scale STM images. An example is shown in Fig. 2d. In the first layer [BMP][TFSA] forms a homogeneous layer across the whole surface. Island formation in the submonolayer coverage range, as it was reported on Au(111),9 Ag(111)10 and Cu(111),12 could not be identified in the present work, not even at temperatures of 80 K, which is the lowest temperature accessible for our setup. Hence, attractive molecule–molecule interactions must be weaker on TiO2(110) than on the metallic substrates mentioned above, or are even absent.
Finally, we also did not observe any preference for adsorption at the TiO2(110) steps (structural effects), whereas this was commonly observed for metallic substrates. In the latter case island growth started preferably from the steps, in the case of Au(111) also at the elbows of the herringbone surface reconstruction.11
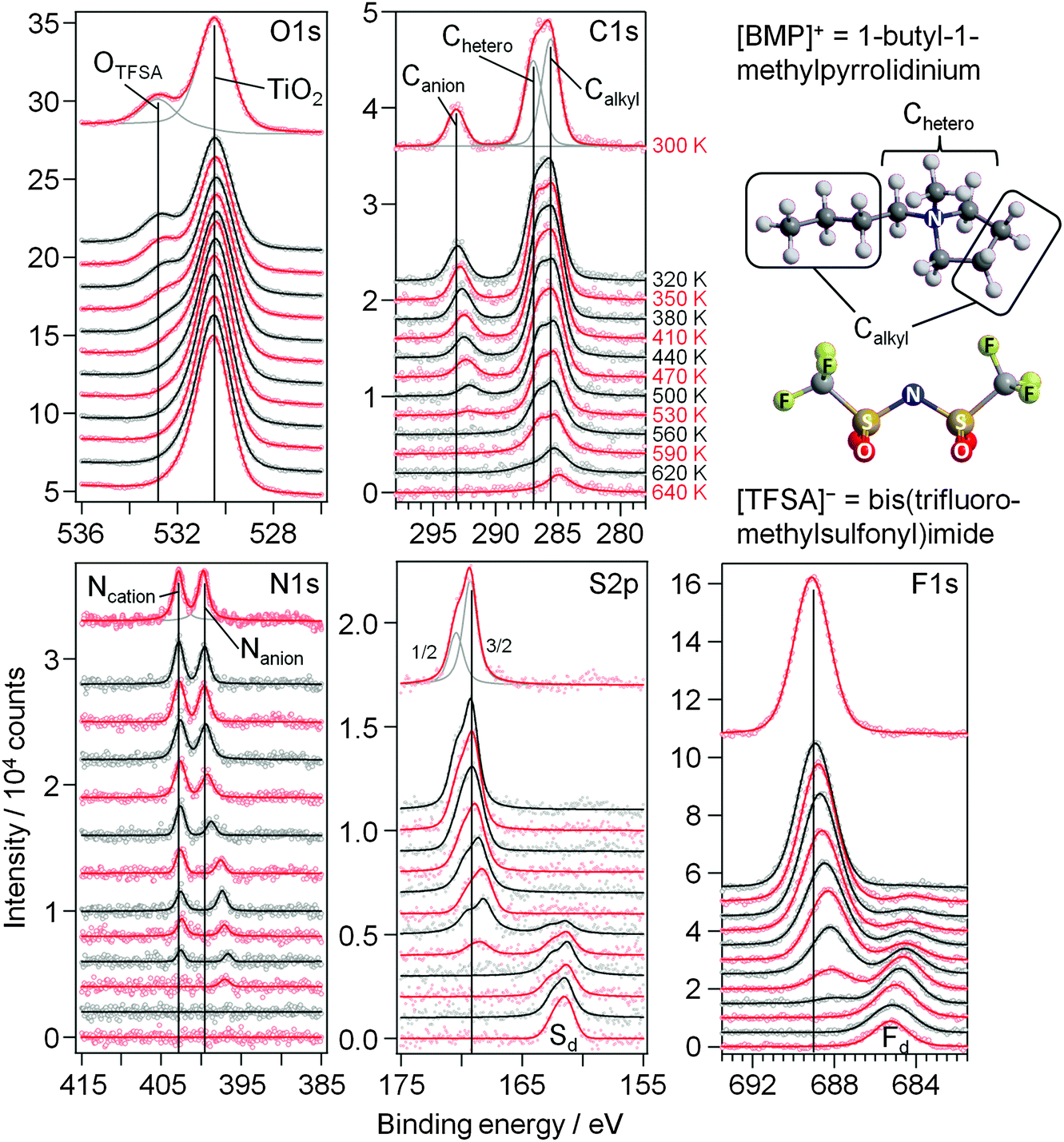 | ||
| Fig. 3 XP core level spectra of 1.5 ML [BMP][TFSA] adsorbed on TiO2(110) recorded upon stepwise heating from 300 K to 640 K in steps of 30 K. The structural model of [BMP][TFSA] is displayed on the right hand side. | ||
In the F1s region the original [TFSA]− related peak shifts successively by 0.9 eV to lower BE between 300 and 500 K and concomitantly decreases in intensity. At 380 K a new peak starts to grow at 684.6 eV, accompanied by a further intensity decrease of the [TFSA]− related peak. The BE of the new species corresponds to signals measured for adsorbed F− atoms on TiO2(110).47–49 We would like to note that CF3-containing molecules are known to decompose via generation of F− under reductive conditions.50 For thermal decomposition of [BMP][TFSA] on Cu(111) no such peak was observed.28 Between 380 K and 640 K, this peak shifts by 0.6 eV to higher BE. At this point we can only speculate that this shift is either due to a change in the adsorption sites with higher temperature or an incorporation of the F− ions into the subsurface region of the TiO2(110) crystal. The [TFSA]− related peak vanished completely at 560 K.
In the C1s region the Canion related peak follows the trend of the other anion signals, with a shift of 1.0 eV to lower BE and a continuous decay of the peak intensity until it vanished at 560 K. The signals from the cation carbon shift only by ∼0.3 eV and persist up to higher temperatures compared to the anion, up to 590 K for the Chetero and up to 640 K for the Calkyl related peak. In addition, the intensities of these signals decrease slower in intensity than all other signals including the Ncation signal. Since the BE of ∼285.3 eV is characteristic also for various kinds of carbonaceous species, we expect that the slower decrease in intensity is related to the formation of carbon containing decomposition products. Since no corresponding signal is found in the N1s region, these compounds do not include any nitrogen any more. A more accurate prediction of the species formed during decomposition is not possible from these XPS measurements (see also the later discussion). Starting at temperatures of ∼410 K, the intensity of the Ccation peaks decays slower than that of the other XP signals during heating, indicating that carbon containing adsorbed decomposition products are formed, while for the other elements the fraction of volatile (desorbing) decomposition products is higher. This discrepancy reaches its maximum at 530/560 K, where the decomposition products account for roughly 80% of this peak-multiplet. At even higher temperature the carbon containing decomposition products start to desorb as well, either directly or via further decomposition followed by desorption, and the related signals have essentially vanished at 640 K.
The above data in combination indicate that multilayer desorption occurs between 300 K and 350 K. This is followed by decomposition of both cation and anion species, accompanied by desorption of decomposition products and possibly also of intact cation and anion species. Decomposition starts at ∼380 K with the formation of adsorbed F− and hydrocarbon species. Since the overall peak intensities in the F1s and S2p regions decrease above this temperature, we assume that only part of the [TFSA]− anions decomposes, leaving adsorbed decomposition products, while the other part desorbs fully, possibly as intact molecules. At 440 K the amount of decomposition products increases more strongly, and Sad is formed in addition, which replaces bridge-bonded surface oxygen in the substrate. Finally, nitrides are also formed on the surface, presumably as TiNx species. At 530–560 K, [BMP][TFSA] is completely decomposed. Upon further heating up to 640 K, also the signals of the TiNx and of the hydrocarbon decomposition products vanish. In the case of TiNx we expect that these species dissolve into the bulk TiO2 or decompose to N2 and desorb. The adsorbed carbonaceous species are expected to further decompose and desorb. The Sad and Fad species finally remain on the surface.
A similar decomposition pattern of the [TFSA]− anion was also found for [HMIM][TFSA] (1-hexyl-3-methylimidazolium [TFSA]−) on reduced CeO2−x surfaces.51
The decomposition of [BMP][TFSA] on TiO2(110) takes place over a wide temperature range, involving various and changing reaction intermediates/products. This points to a complex multistep mechanism, which is presumably influenced also by the structure of the adsorption site (kink site, different step sites, terrace position). To gain further insight into the decomposition behaviour, further input from experiment and theory is required, such as high resolution STM data recorded at even lower temperatures or quantum chemical calculations. So far, the decomposition of the [BMP]+ cation under reductive electrochemical conditions was investigated in DFT-based calculations,52 and the reductive electrochemical decomposition of the [TFSA]− anion in [PMP][TFSA] (PMP = n-propyl-n-methyl-pyrrolidinium) was explored by ab initio calculations.53 Furthermore, the thermal decomposition of [BMIM][TFSA] (b = butyl) was mapped out via DFT based calculations.54 Finally, the thermal decomposition of bulk [BMP][TFSA] was characterized by thermal gravimetric analysis, coupled with mass spectrometry detection of the reaction products (TGA-MS).55 The most likely pathways calculated for the decomposition of [TFSA]− are shown in the upper part of Fig. 4a. For the electrochemical decomposition (left side), one of the S–N bonds breaks in the first step, induced by the uptake of one electron at negative electrode potential. The resulting SO2CF3− and NSO2CF3− species further react to SO2(−), NSO2(−) and CF3− by breaking the S–C bonds, and they may further decompose to sulfide, oxide, fluoride and carbide anions.53 For the thermal decomposition (right side), DFT calculations were performed for isolated molecules, where no electron-transfer from the outside is possible. The energetically most favourable decomposition pathway under these conditions proceeds via the release of sulfur dioxide.54 Considering the very different environment in these calculations, one has to be careful when transferring those results to the present experimental findings. Nevertheless, the calculations provide valuable information on the possible decomposition products on the TiO2(110) surface. Both decomposition pathways (see Fig. 4a) have in common that they break the same bonds in the molecule, therefore they are most likely also the ones which will be broken upon decomposition of [BMP][TFSA] on TiO2(110).
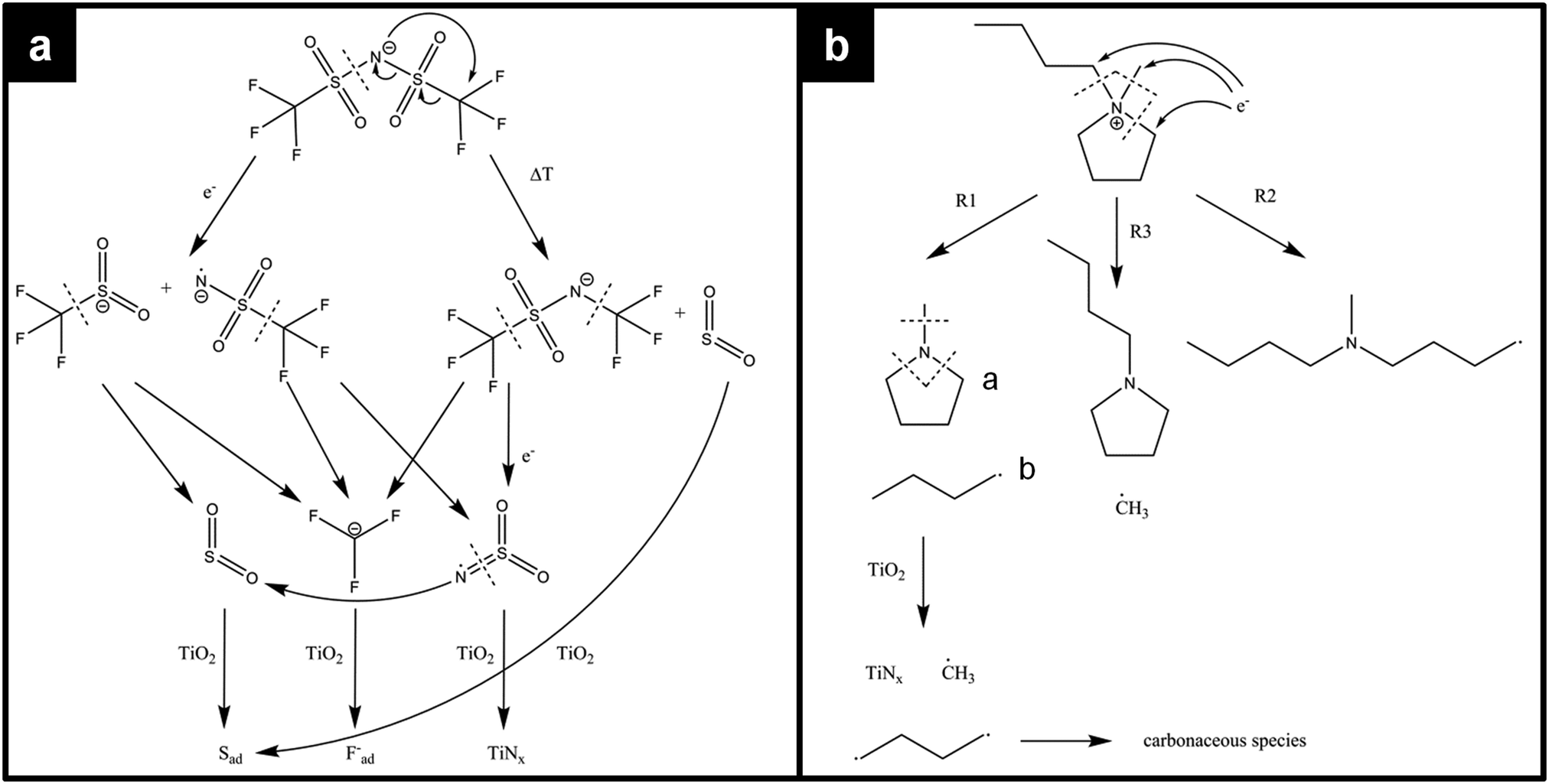 | ||
| Fig. 4 Possible decomposition mechanisms of (a) the [TFSA]− anion and (b) the [BMP]+ cation according to calculations of ref. 52–54 and thermal decomposition products found on the TiO2(110) surface according to our XPS measurements. | ||
DFT calculations on the decomposition of a [BMP]+ cation (under reductive electrochemical conditions) revealed three possible reaction pathways R1–R3, which are shown in Fig. 4b.52 R1 is the energetically most favoured one, followed by R2 and R3. The TGA-MS measurements (see above) showed that thermal decomposition of [BMP][TFSA] occurs at above 673 K with MS signals at 41, 56, 85 and 69 amu in the gas phase above the solvent, which were attributed to C3H5+˙, C3H6N+˙, and C5H11N+˙ fragments from the cation and CF3+˙ from the anion. These results agree with the calculations insofar as in both cases CF3 species are formed. On the other hand, SO2 was proposed as a decomposition product based on the calculations, but was not detected in the TGA-MS measurements. This discrepancy may arise from a too low volatility of these species, such that they are not vaporized and remain in the liquid (possibly as SO3−). The C5H11N+ fragment fits to molecule “a” formed by reaction pathway R1 in Fig. 4b. The fragment C3H5+˙ most likely belongs to a butyl-species like molecule “b”, which is also formed in pathway R1. The butyl species is cracked in the ionization zone of the mass spectrometer (among other reactions) into a C3 and a C1 fragment, where the C1 fragment is uncharged and therefore not detected. This decomposition pattern was concluded from comparing the mass spectrum of n-butane, where amu 41 and 42 signals are among the most intense signals.56 Comparing the DFT calculations for electrochemical decomposition with the TGA-MS measurements, it is likely that the decomposition of bulk [BMP][TFSA] proceeds along the reaction pathway R1. Coming back to [BMP][TFSA] on TiO2(110), thermal decomposition does not stop at the stage described in the three reaction mechanisms. Based on our XPS results, the methyl-pyrrolidinium species is presumably decomposed by breaking the N–C bonds, forming butyl and CH3 species. The nitrogen (ad)atoms react with the rutile surface forming TiNx, which is detected in the XP spectra. If butane is formed, this is likely to desorb at lower temperatures than detected here, since for other surfaces desorption temperatures of <200 K were reported.57–59 Therefore we assume that the adsorbed reaction products detected by XPS are fragments of the butyl species formed in the decomposition process of [BMP]+. The adsorbed reaction products should solely contain C (and H) atoms based on the XPS results. A more detailed assignment is not possible, since a BE of around 285 eV is typical for a wide variety of CHx containing species, and even graphite shows a peak at around 284.7 eV.60,61 On the other hand, the [TFSA]−ad intermediates are further decomposed: for SO2 it is known from the literature that it forms sulfite (SO32−) and sulfate like species by attachment of SO2 to the bridge-bonded oxygen species of the TiO2(110) surface in a temperature range between 135 K and 450 K.62,63 For TiO2(110) with a high density of surface defects (e.g., after sputtering) or with a high step density (e.g. TiO2(441)), SO2 was reported to react to Sad even at room temperature.63–65 Our observation of Sad formation at temperatures >500 K, but no formation of SOx, can be explained the following way. The SO2 generated by the decomposition of [TFSA]− forms adsorbed sulfite and sulfate species. At the high temperatures these may instantaneously react to Sad, or the decomposition of the [TFSA]− creates a high density of defects on the TiO2(110) surface, which catalyse the reaction SO42− → Sad. If adsorbed CF3 and NSO2 species are formed in the decomposition process of [TFSA]−, these are also decomposed forming Fad, TiNx and SO2, where the latter again reacts to Sad, which are detected in the XPS measurements. The remaining carbon may finally further react to CO or CO2, which both desorb instantaneously.18
2.2 Interaction of Li with TiO2(110)
First we present results of XPS measurements on the interaction of Li with TiO2(110). Detailed spectra of the Li1s, Ti2p and O1s regions are shown in Fig. 5a. The spectra were recorded after stepwise deposition of Li on a clean TiO2(110) surface (topmost spectra) at 80 K. The bottom spectra show the highest Li dose (corresponding coverage ∼9.0 ML Li). In the Li1s region a peak emerges at 57.2 eV after the first Li dose (0.5 ML). Upon increasing the amount of deposited Li, this peak grows in intensity and shifts lower BEs, to 56.4 eV at 9.0 ML. Generally, the intensity of the Li1s peak is very low due to the very small atomic sensitivity factor of this peak. Interestingly, the peak positions are at significantly higher BE than for bulk Li (54.8 eV66) or for Li adsorbed on Cu(111) (55.1 eV28) and even higher than reported for intercalated Li in anatase22 or LTO.67 Therefore we assume that Li has reacted with the surface and exists as a Li+ species, either as a LixO species at the surface or intercalated into the substrate, rather than being atomically adsorbed on the surface.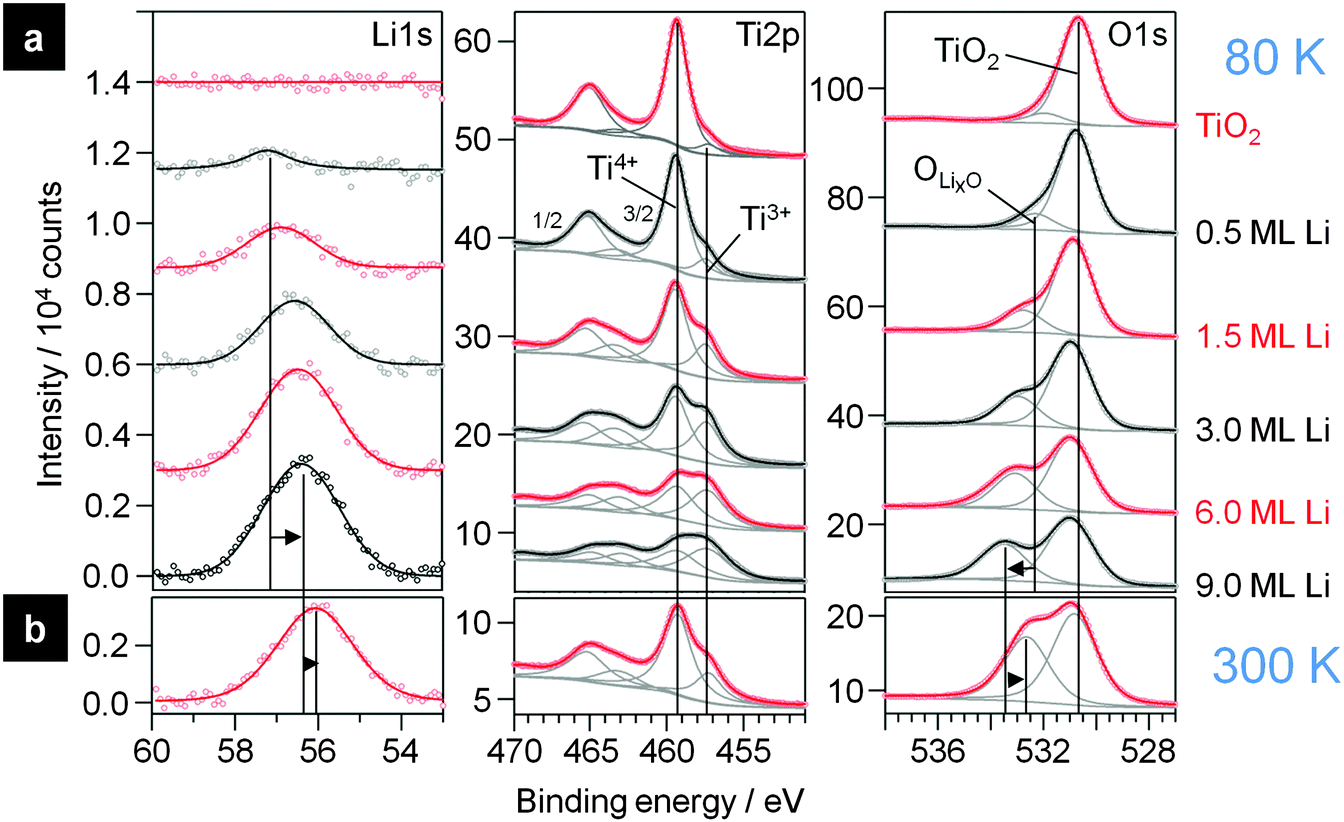 | ||
| Fig. 5 (a) XP detail spectra of the Li1s, Ti2p and O1s region of clean TiO2 (topmost spectra) and upon increasing evaporation of Li onto this surface at 80 K. (b) XP spectra after slowly heating up the sample overnight to 300 K. | ||
This assumption is supported by the corresponding Ti2p and O1s spectra, where new peaks are formed upon deposition of Li. After the first Li dose (0.5 ML), an additional new doublet evolves in the Ti2p region at 2.0 eV lower BE than the doublet of the initial Ti2p state of TiO2(110). In the O1s region also a new peak (“OLixO”) appears, which is shifted by 1.5 eV to higher BE relative to the TiO2(110) peak. The new OLixO peak shifts to higher BE with increasing Li deposition, by 1.1 eV at 9.0 ML. This is very similar to the results obtained for Li intercalation in anatase at RT, where electrochemical insertion of Li also caused an additional Ti2p peak at lower BE relative to the original TiO2 peak.22,24 Note that in our measurement these additional peaks appear already with weak intensities in the spectra of the clean rutile sample. We explain this by small amounts of Ti3+ species in the crystal due to the reductive pre-treatment (Ti2p state) and to residual contaminations from previous experiments with Li on the same crystal. Nevertheless, the evolution of these peaks clearly demonstrates that the atomic Li reacts with the surface upon interaction with Li even at 80 K. The additional doublet in the Ti2p region is attributed to the formation of Ti3+ species, which are formed by the transfer of one electron from Li0 to a Ti4+ species in the rutile crystal. Since there is also a second peak in the O1s region evolving at lower BE relative to the original TiO2 peak, we expect that the resulting Li+ atoms are positioned next to O2−-atoms in the crystal which therefore have a different electronic surrounding. Another important question is whether this reaction takes place solely on the surface or if intercalation into the near surface region or bulk rutile crystal occurs as well. For this purpose we concentrate on the changes of the peak intensities. In the spectra recorded on pure TiO2(110) the Ti2p:O1s peak intensity ratio (corrected by the sensitivity factors) is 0.54 ± 0.02, which is slightly higher than the expected ratio of 1:2. We tentatively explain this by the presence of oxygen vacancies and Ti3+ species. Upon addition of Li this ratio is decreased to 0.32, because the intensity of both O1s peaks together increases to 123% while the total intensities of the Ti2p peaks fades to 69% compared to the pure rutile signals (see below). The intensity ratio between the Ti4+ and Ti3+ peaks is very close to 2:3. The high fraction of reduced Ti3+ species implies that these are not only in the surface layer, but also in deeper layers, meaning that diffusion of Li into the subsurface regions is possible under these conditions and leads to the formation of Ti3+ and Li+ in these regions. The ratio of 2:3 would fit to the stoichiometry of a Li loaded LTO spinel Li7Ti5O12. In LTO the ratio between Ti and O atoms is smaller than in rutile TiO2, therefore part of the lower relative intensity of the Ti2p peaks, relative to that of the O1s signals, can be explained by the change in stoichiometry from Ti:O of 1:2 (or 0.5) in rutile to 5:12 (or 0.417) in LTO. The measured Ti:O ratio of 0.32, however, is even lower, in agreement with the existence of a Li oxide (LixO) cover layer on top of the LTO-like layer. In that case the peak at higher BE in the O1s region must originate from two different species: the LixO formed on top of the substrate and oxygen species in the subsurface region which are neighboured to intercalated Li+. Based on the higher Li1s BE (bulk Li2O: Li1s: 53.8 eV; O1s: 528.5 eV68,69), the LixO layer must be electronically different from bulk Li2O.
The calculation of the thickness of the LixO overlayer is not simple since the decay of the Ti2p peak intensity originates from both the LixO cover layer and the change in stoichiometry of the TiO2(110) substrate in the near surface region. The same combination of effects is responsible also for the higher intensity of the O1s signal (123%) compared to the XPS signal of the Li free sample, and for the evolution of the Li1s peak, which originates both from the Li in the LixO surface layer and from the Li in the LTO like layer. For an estimate of the LixO layer thickness we assume that the Ti2p signals are generated by a LTO layer and calculate the O1s signal which corresponds to this Ti2p signal to achieve the stoichiometry between Ti and O in LTO. Subtraction of this value from the more intense measured O1s signals results in the oxygen fraction which originates from the LixO layer. Details of the calculation can be found in the ESI.† For deposition of 9 ML Li this calculation results in a thickness of the LixO cover layer of ca. 0.1–0.2 nm, which is ≤1 ML of LixO.
The thickness can also be calculated from the damping of the O1sLTO or of the Ti2p signals,70 where the latter is again assumed to fully result from a stoichiometric LTO layer (details on the calculation are found in the ESI†). Both estimates give a result for the thickness of the LixO layer which is in the same range as it was found in the first calculation. The assumption that a fully stoichiometric LTO substrate is used for the calculation appears plausible for the sample with 9 ML Li deposited and an detection angle of 80°, since in this case the layer with LTO like stoichiometry is expected to be clearly thicker than the detection depth (details on the calculation of the LTO layer thickness are given in the ESI†).
The small thickness of the LixO layer is presumably the reason why the peaks are shifted to higher BE compared to bulk Li2O because of the interaction to the rutile crystal underneath. Furthermore, it may also be that for these low coverages no homogeneous, closed layer of LixO is formed but instead oxygen and Li+ are enriched on/in the first surface layer. The mobility of the Tin+ ions in rutile seems to be high enough that diffusion is possible from the bulk TiO2(110) in the near surface region and to the surface even at 80 K. The Li:O stoichiometry in this LixO film cannot be quantified precisely because of the low intensity of the Li1s peak, leading to problems in the background subtraction, and since the Li1s peaks of the Li species at the surface and those intercalated in the near surface region appear at the same BE.
To elucidate how thick the LTO layer between the LixO layer and the bulk rutile is we also conducted the same XPS measurements as shown in Fig. 5a with a detection angle of 0° with respect to the surface normal (the spectra are shown in the ESI†). Here the mean escape depth is with ca. 2.9 nm higher than for 80° with 0.5 nm. For these signals the Ti4+ peak nearly has double the intensity compared to the Ti3+ peak at the highest investigated Li loading. Therefore we assume that now a mixture of the LixO layer, followed by Li7Ti5O12 and presumably a layer with a decreasing Li concentration on top of unchanged bulk rutile is probed. Since we cannot derive information on the type of the concentration gradient of Li+ and Ti3+ from our XPS measurements, because we are always sampling the average of the uppermost atomic layers (and a deep profiling with sputtering is also not possible since this would alter the surface), we cannot determine the thickness of the LTO-like layer except for the fact that for 9 ML Li deposition it is thicker than the sampling depth of the measurement at 80°. Nevertheless, for a rough estimate we calculated the thickness assuming a layer geometry of LixO on top of LTO on top of TiO2(110) with no concentration gradients in between. Under these conditions the LTO layer thickness is calculated to be 2.0 ± 0.1 nm. Upon slow warm up of the sample to 300 K overnight the stoichiometry changes. Spectra recorded at the 80° emission angle after annealing are shown in Fig. 5b. The overall damping of the Ti2p peaks stays the same, indicating that the LixO layer is still present, but the intensity ratio of the Ti4+:Ti3+ peak changed to ∼5:1 (2.7), indicative of a distinct migration of Ti3+ species deeper into the bulk. For measurements conducted at the 0° emission angle the relative intensities changed marginally from (Ti4+ + Ti3+):O = 0.42, Ti4+:O = 0.33 and Ti3+:O = 0.08 at 80 K to (Ti4+ + Ti3+):O = 0.43, Ti4+:O = 0.36 and Ti3+:O = 0.06 at RT. This means that in the uppermost 1–2 nm, which are probed at the 80° emission angle, the composition changes strongly, while the average stoichiometry in the uppermost ∼5 nm stays about the same. The higher temperature enhances the diffusion into the bulk and the Li+ and Ti3+ species agglomerated in the first 1–2 nm at 80 K move deeper into the material at higher temperature. The Li7Ti5O12 layer in the surface region therefore was kinetically stabilized by the low temperatures and upon heating the gradient of Li+ and Ti3+ interstitials is reduced. Nevertheless, since the signal at 0° detection angle changes very little, the Li ions seem to remain in the layer probed here, in the topmost 5 nm.
To summarize our XPS results on Li deposition on TiO2(110), we found that even at 80 K part of the deposited Li intercalates, while the remaining part forms a thin adlayer of LixO like species. Because of diffusion limitations, the Li species stay in the near surface region and form a layer with a LTO-like stoichiometry, as evidenced by the concentration of Ti3+ species. At higher temperature (300 K), diffusion becomes faster and the Li+ and Ti3+ species are probed not mainly in the first 2 nm, resulting in a lower signal intensity of these species at the 80° detection angle in XPS. Since the signal intensities did not change significantly for detection at 0°, the Li+ and Ti3+ species are still in the uppermost ca. 5 nm which are probed in this geometry.
Comparing the present XPS results on Li interaction with rutile TiO2(110) with previous related findings for Li electrodeposition, but also for Li deposition in UHV on TiO2, both for anatase and for rutile, leads to the following picture: investigating the insertion of Li into thin films of anatase with XPS (synchrotron radiation as the X-ray source) at room temperature, Henningsson et al.22,23 found a new peak in the Ti2p region growing simultaneously with the successive deposition of Li, which appeared at 1.6 eV lower BE compared to the original Ti2p peak related to Ti4+ species in the substrate. For high amounts of Li (>5 ML) a third peak emerged at even lower BE. These two peaks were attributed to the formation of Ti3+ and Ti2+ species in the substrate upon Li insertion into the material. In the Li1s region they observed peaks at 56.5 eV and 55.5 eV with the peak at higher BE forming at coverages up to 2.8 ML. The peak at lower BE appears at coverages >0.64 ML and dominates the spectrum above 2.8 ML. The authors explained these results by the formation of two phases in anatase, with a Li rich Li0.4TiO2 phase and a Li poor Li<0.07TiO2 phase. This is in agreement with findings for the electrochemical insertion of Li into anatase.26 Södergren et al.24 combined electrochemical Li insertion with XPS measurements and found that the formation of the Ti2p peak related to Ti3+ is reversible when cycling the sample potential, which shows that the original anatase structure is recovered upon deintercalation of Li. Comparison with the studies mentioned before indicates that electrochemical Li insertion and Li insertion upon evaporating Li in UHV on anatase proceed in a comparable process. Li deposition on anatase TiO2 allows rapid Li intercalation at room temperature, also under the conditions applied in the present study (Li evaporation in UHV).
On rutile TiO2, intercalation of Li is more difficult. For bulk materials it was thought for a long time that at RT Li intercalation is not possible at all. Zachau-Christiansen et al. reported that the electrochemical Li insertion into rutile was inhibited at 25 °C, only for reaction at 120 °C two Li containing phases with stoichiometries of Li0.13–0.15TiO2 and Li0.5TiO2 were observed,26 comparable to observations for anatase (see above). More recent studies, however, indicated that intercalation is feasible also at RT, e.g., by reducing the size of the rutile particles, e.g., to needles with diameters of 5–20 nm.25 This was explained by a slow diffusion of Li at 25 °C, especially perpendicular to the c channels in the crystal. (The c channels run along the [001] direction in the (110) lattice plane, therefore intercalation into a TiO2(110) terminated crystal has to proceed along the slow diffusion direction.)
These results are in good agreement with our findings when keeping in mind that the earlier studies on rutile mentioned above were conducted determining the bulk composition of the TiO2 materials rather than surface sensitive techniques. Using surface sensitive measurements we could demonstrate that Li intercalation starts already at much lower temperatures. Under those conditions it is limited, however, to the near surface regions because of the slow diffusion of the Li species. Our results agree also with the findings by Krischok et al.,20 who used MIES (metastable impact electron spectroscopy) and UPS (HeI) to study exactly the Li interaction with rutile TiO2(110) at temperatures between 130 K and 620 K, where in particular MIES is even more surface sensitive than XPS (although it does not allow to quantify how much and how deep the Li is inserted). They reported that at 130 K Li adsorbs ionically up to 0.3 ML and Ti3+ is formed in parallel. At higher coverages the ionic character of the adsorbed Li atoms is smaller (e.g. at 0.7 ML Li0.4+). Above 160 K, the Li2s signal in the very surface sensitive MIES spectra disappeared almost completely, which they explained by (almost complete) Li insertion into the bulk. This agrees at least qualitatively with our findings, when using comparable Li coverages of 1.4 ML.
In addition to XPS we performed STM measurements of the surface after evaporating Li at 80 K. Representative STM images are shown in Fig. 6. The first image, recorded after 1.5 ML Li deposition, provides an overview of the surface morphology after deposition, while the smaller scale image in Fig. 6b resolves also details of the surface structure. The steps and terraces of the substrate seem to be unchanged after Li deposition. On the surface we find a number of protrusions which are (nearly) uniformly spread across the surface. At some places also the original TiO2(110) surface with its 1 × 1 structure is visible in Fig. 6b (two regions are marked by red circles). The round protrusions may correspond to LixO species, which according to our XPS data are formed upon Li deposition on top of the substrate. Another possibility would be that they result from the change of the substrate itself, reflecting the formation of a LTO like species. The STM images do not indicate any change in appearance between 80 K and RT. Comparing our results with the STM data reported by Tatsumi et al. for room temperature adsorption of Li on TiO2(110), our STM images correspond to those shown in their work for higher deposition times (600 s in Fig. 2b in ref. 21). Note that the latter authors assigned the new structures observed in their STM images to Li adatoms, which seems to be in contrast to our findings and to those of other authors.20 We rather expect that these structures reflect the initial stage of Li insertion and formation of LixO. Since the protrusions detected are mostly situated on the rows of 5-fold coordinated Ti4+ species,21 one may speculate that they are generated by Ti3+ species formed by the reaction with an impinging Li atom or by an adsorption complex of Li+ on Ti3+.
 | ||
| Fig. 6 Large (a) and small scale (b) STM images of a TiO2(110) surface after Li deposition at 80 K. At some places (e.g., marked by red circles) parts of the TiO2(110) structure are still visible (a: T = 108 K, UT = 1.1 V, IT = 21 pA; b: T = 102 K, UT = 1.1 V, IT = 21 pA). | ||
Furthermore we may compare our STM images with AFM images of single crystalline LTO(111) surfaces reported by Kitta et al.,71 who performed AFM measurements on surfaces which were electrochemically charged by Li insertion. They observed a surface roughening, and their AFM images at the initial stage of the Li insertion are very similar to our STM images. This seems to indicate that the initial Li insertion in TiO2(110) occurring upon Li deposition under UHV conditions closely resembles the initial stage of electrochemical Li insertion into LTO.
2.3 [BMP][TFSA] + Li on rutile TiO2(110)
This section focuses on the interaction of coadsorbed [BMP][TFSA] and Li with each other and with the TiO2(110) substrate. First we discuss the effect of Li deposition on a TiO2(110) surface which was pre-covered with 0.7 ML of [BMP][TFSA]. XP core level spectra recorded after Li deposition at 80 K are shown in Fig. 7. From top to bottom spectra recorded on the clean TiO2(110) substrate (a), after deposition (80 K) of 0.7 ML of [BMP][TFSA] (b), and after stepwise deposition of Li (c–h) are presented. In the Ti2p region deposition of [BMP][TFSA] causes a damping of the bulk TiO2 (Ti4+ and Ti3+) peaks. With increasing Li uptake (c–h) the intensity of the Ti4+ decreases further, and that of the Ti3+ signals increases, comparable to the findings for Li deposition on pure TiO2(110), without [BMP][TFSA]. Obviously, also under these conditions Li reacts with the TiO2(110) substrate and intercalates into the substrate. This is confirmed by an additional peak in the O1s spectral range (OLixO), which appears at a BE of 532.3 eV between the two peaks OTFSA and OTi2O and shifts gradually to 532.7 eV with increasing Li deposition. This peak is at the same BE as the additional peak for Li deposition/intercalation on/in TiO2(110) in the absence of a [BMP][TFSA] adlayer, and therefore attributed to the same species (O species with changed electrochemical surrounding due to the neighbouring Li+ species). The intensity of the O1s rutile peak stays (nearly) constant during Li deposition, while that of the OTFSA peak strongly fades to 37% of its starting value and shifts by 1.1 eV to higher BE. The intensity decay of the [TFSA]− related peaks implies that the deposited Li atoms not only react with and intercalate into the TiO2, but also partly react with the adsorbed [BMP][TFSA], even at low temperature. Therefore the OLixO peak could at least partly result from decomposition products of the IL anion. Possible reaction products would be SOx or SOx containing species, since according to the literature SO32− adsorbed on TiO2(110) has roughly the same BE, between ca. 532 and 533 eV (for related S peaks see below).64 When comparing, e.g., the peak intensities of Ti2pTi4+, Ti2pTi3+ and O1sLixO signals in this measurement, using the highest Li deposition of 6 ML, with those obtained for a similar Li uptake on a pure TiO2(110) substrate, the intensity ratios of Ti4+:O1sLixO (0.27) and Ti3+:O1sLixO (0.16) are much lower for TiO2(110) + IL + Li than for TiO2(110) + Li (0.53, 0.5). Since the intercalation into the TiO2(110) surface region should result in (roughly) the same stoichiometry for the same Li dose/same temperature, the O1sLixO peak is too intense relative to the Ti2p peaks to only originate from Li intercalation. Therefore we expect this peak to include also contributions from [BMP][TFSA] decomposition products of the SOx type. For the highest amount of Li deposition (6 ML) the two species should contribute about equally. The formation of a SOx species is supported also by a new peak in the S2p regime. Here an additional peak doublet (marked as Sd1 in Fig. 7) increases with Li deposition at BEs of 167.5 (S2p3/2) and 168.7 eV (S2p1/2), which shift to 169.1 and 170.4 eV, respectively, with increasing Li deposition. This fits well to SO32− or SO42− species adsorbed on TiO2(110)64,72–74 and Li2SO3.75 Nevertheless, considering also the peaks in the other regions (see also discussion below), this peak also must include contributions from F3C–O2S–N−Li+ species. In addition to the Sd1 peak, two new peak doublets develop with higher Li amounts at lower BEs of 166.0 and 162.8 eV (position of the S2p3/2 peak of each doublet). The latter one is at 1.5 eV higher BE than the peak obtained upon thermal decomposition of a pure [BMP][TFSA] adlayer on TiO2(110), which was attributed to the formation of Sad. Accordingly, this may be of similar origin in the present case, but with a slightly higher BE due to a different chemical surrounding. It is also possible, however, that a LixS species is formed, whose S2p3/2 signal is expected to appear in the same range of BEs (162.0 eV).28 The origin of the other S2p3/2 peak at 166.0 eV is not fully clear. Its BE is too low for a SO32− or SO42− species,64,73,74 but it could be due to a F3C–O2S−Li+ like species following the reaction scheme for [BMP][TFSA] decomposition in Fig. 4a. The original STFSA peak has lost almost 85% of its initial intensity after the highest Li deposition. This is much more than the loss of the OTFSA peak, which was about 63%. Therefore also the OTFSA peak must contain contributions from decomposition products with similar BE. Possible candidates would be F3C–O2S–N−Li+ and F3C–O2S−Li+. The same is true for the N1s region, where the two peaks originating from intact [BMP][TFSA], Ncation and Nanion, lose about 2/3 of their initial intensity during Li evaporation and shift by 1.9 and 1 eV to higher BE, respectively. Due to the large shift of the Ncation peak this peak is expected to also contain contributions from CxHyNz species, which appear at similar BE’s. Finally, another N1s peak appears at BE 398.7 eV, 2 eV higher than the additional peak observed upon thermal decomposition of [BMP][TFSA] on TiO2(110), which we had tentatively attributed to TiNx. Therefore the new signal at 398.7 eV must be due to another reaction product. Signals at this BE were attributed to Li3N species in a postmortem XPS analysis of Li-ion batteries,75 or to reaction products arising from the interaction of [OMIM][TFSA] with Li on a polycrystalline Cu foil27 and [BMP][TFSA] with Li on Cu(111).28
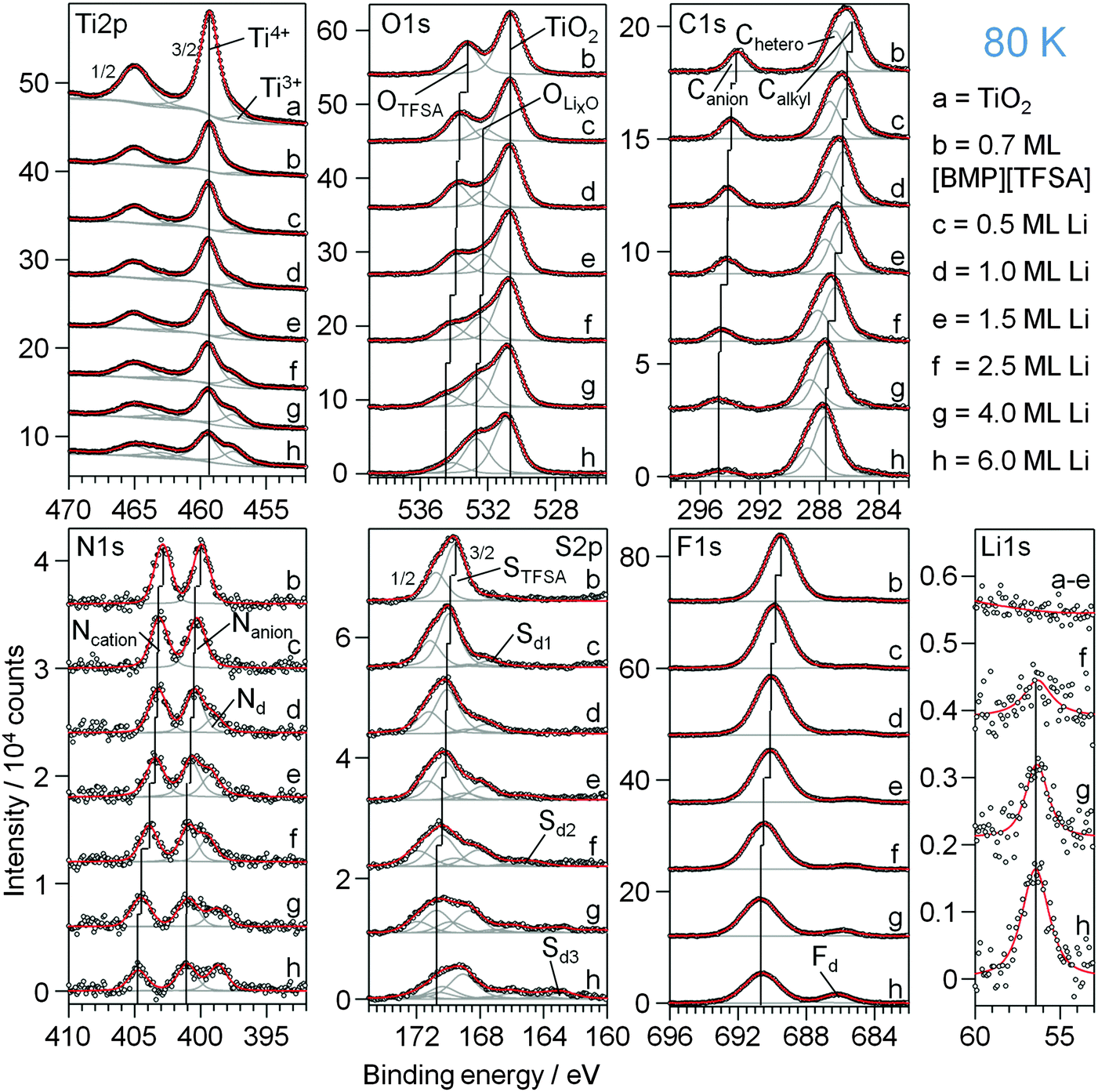 | ||
| Fig. 7 XP detail spectra of (a) a clean TiO2(110) surface, (b) covered with 0.7 ML [BMP][TFSA] and (c–h) after increasing amounts of Li post-deposited step by step onto [BMP][TFSA]|rutile(110); the sample was always at 80 K. | ||
In the F1s region the original FTFSA peak at 689.5 eV decreases to 50% of its initial intensity and shifts by 1.1 eV to higher BE during Li deposition, while at the same time a new peak grows in at 685 eV, which shifts by 1.2 eV to higher BE with increasing Li coverage. This peak is attributed to adsorbed F− species, similar to the assignment made for thermal decomposition of [BMP][TFSA] on TiO2(110), and to the formation of LiF.75
Finally, in the C1s region, no new peaks are formed in addition to the initial [BMP][TFSA] peaks, but the Canion peak decreases to ∼1/3 of its initial intensity and shifts by 1.1 eV to higher BE upon Li deposition, whereas the Chetero peak loses only 20% in intensity together with a shift by 1.8 eV to higher BE. The Calkyl peak even grows to 146% of its initial intensity, together with a shift by 1.8 eV to higher BE. Based on these data, the FTFSA and Canion peaks are expected to contain contributions from F3C–O2S–N−Li+ like reaction products. For the C1scation peaks, possible reaction products include various CxHyNz and CxHy species, which cannot be identified in more detail.
Comparing the decomposition products of the [TFSA]− anion and their formation found here by XPS with the decomposition mechanism predicted for reductive electrochemical decomposition of [TFSA]− by Howlett et al.53 (also summarized on the left hand side in Fig. 4a), it is found that they are in principle identical. This can plausibly be rationalized by assuming that the impinging Li atoms readily donate their valence electron to form Li+, which is comparable to the reductive electrochemical conditions employed by Howlett et al.53
For comparison we also deposited Li on a TiO2(110) surface covered by a multilayer film of [BMP][TFSA] (4.7 ML). The spectra are included in Fig. S2, ESI.†. In this case, no Ti3+ species are formed, and therefore Li intercalation can be excluded. Instead, Li only reacts with the [BMP][TFSA] film. The decomposition products are identical with those observed for the reaction with [BMP][TFSA] at submonolayer coverage. In particular the peak which was related to OLixO before is formed as well. In this case, however, it does not stem from LixO, but from decomposition products of the [TFSA]− anion such as F3C–O2S–N−Li+ and Li2SO3–4 (see above). This also supports our above conclusion that this peak originates not only from oxygen species in the TiO2(110) crystal, which are modified by neighbored intercalated Li, but also from these [TFSA]− decomposition products. This measurement furthermore demonstrates that the Li induced decomposition of the [BMP][TFSA] adlayer is not catalysed and not even modified by the TiO2(110) surface. The presence of the substrate mainly affects the decomposition reaction by removing part of the Li via the competing Li intercalation process.
2.4 Thermal stability of a [BMP][TFSA] + Li adlayer on TiO2(110)
The thermal stability of mixed [BMP][TFSA] + Li adlayers on TiO2(110) was investigated by RT deposition of Li (2 ML) on a TiO2(110) surface covered by a submonolayer film of the IL (deposition also at RT) and subsequent stepwise heating of the mixed adlayer covered sample. XP spectra recorded after the respective preparation and heating steps are shown in Fig. 8. As illustrated by the formation of Ti3+ species, Li reacts with and intercalates into the TiO2(110) substrate also upon deposition at 300 K. Furthermore, it also reacts with the IL, and the decomposition products are largely identical with those formed upon interaction at 80 K. Only the Sd2 peak in the S2p regime, which was tentatively related to an adsorbed F3C–O2S−Li+ species, does not appear in the present measurement. Most likely, this species is not stable at this temperature and directly decomposes to SO42−, SO32− or LiSO3 (Sd1).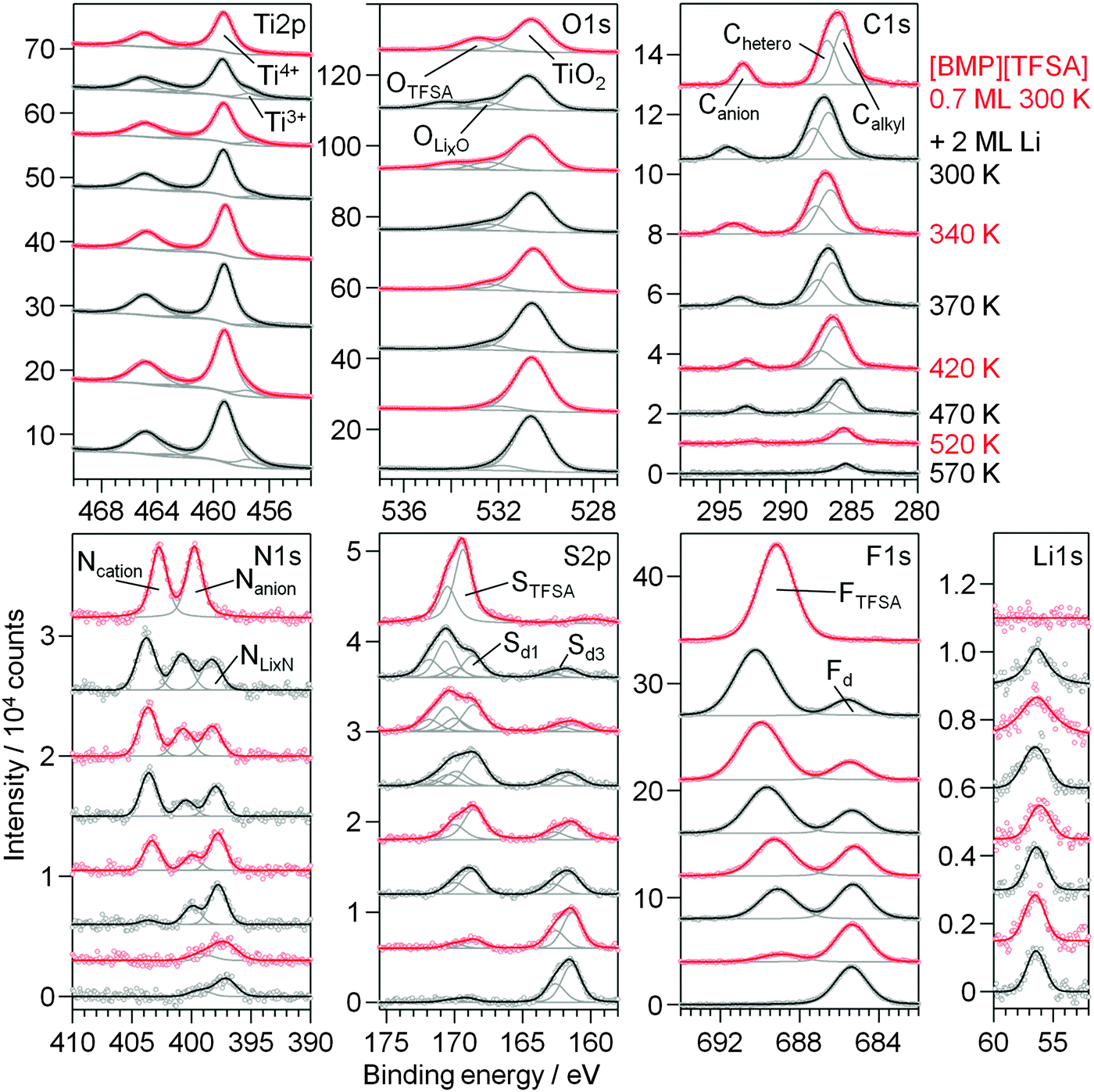 | ||
| Fig. 8 XP detail spectra of 0.7 ML [BMP][TFSA] on TiO2(110) at 300 K (topmost spectra), after post-deposition of 2 ML Li at 300 K and upon stepwise heating to 570 K. | ||
Heating the sample to higher temperatures the decomposition of the adlayer continues (Fig. 8). In the O1s region the peak related to the intact adsorbed [TFSA]− species rapidly loses in intensity between 300 K and 370 K and has completely vanished at 420 K. The same is true for the STFSA signal. Hence, upon annealing to 420 K the intact [TFSA]− is completely decomposed. Therefore the other (anion related) peaks remaining at this temperature must arise from decomposition products. This includes the Canion peak (slightly shifted to a lower BE of 293 eV), the Nanion signal (shifted to a BE of 400 eV), as well as the Sd1, OLixO and FTFSA signals. As discussed for Li deposition on predeposited [BMP][TFSA] thin layers at 80 K, the latter peaks contain contributions also from a [TFSA]− decomposition product, most likely adsorbed F3C–O2S–N−Li+. The Sd1 signal is additionally attributed to Li2SO3–4 and OLixO to LixO. The remaining Ncation peak presumably originates from adsorbed CxHyNz species, following the proposal by Kroon et al. (see Fig. 4b).52 The peak at BE 161.4 eV (marked as Sd3 in Fig. 8) arises due to formation of Li2S. Interestingly, when looking at the Ti2p region, the Ti3+ signal is fading and reaches a minimum at 420 K. It seems that the decomposition reaction of the IL adlayer is induced by reaction with Ti3+ species that were intercalated in the near surface region (first few nm) of the TiO2(110) substrate and start segregating to the surface. Apparently, this goes along with a re-oxidation of the Ti3+ species to Ti4+. Therefore, at this temperature the OLixO peak is no longer (co-)generated by O atoms in the TiO2(110) crystal, but solely due to the decomposition products of the IL. In the Li1s region, there is a small change of the BE from 56.4 eV at 340 and 370 K to 56.1 eV at 420 K. For higher temperatures the Li1s peak shifts back to 56.4 eV and the Ti3+ species forms again. This occurs in parallel to the further decomposition of the adlayer, which is indicated by a strong decrease of the Ncation peak between 370 K and 470 K. We explain these changes to a decomposition of the CxHyNz species related to this peak to H-containing carbonaceous species. At 520 K, also the peaks related to F3C–O2S–N−Li+ have further decreased in intensity, and they completely vanished after annealing to 570 K. At this point also most of the adsorbed carbonaceous species have decomposed and desorbed. The only remaining species are LiF (=Fd), Li2S (=Sd3), LixN (=peak at 399 eV) and TiNx (=peak at 397.1 eV). The remaining OLixO peak is due to the interaction of bulk O atoms with intercalated Li+ (and presumably LixO on the surface).
In a second experiment we tested the stability and decomposition of a mixed [BMP][TFSA] + Li adlayer after the reversed deposition order. In this case Li was deposited on TiO2(110) at 300 K, followed by post-deposition of [BMP][TFSA]. The amount of Li is higher in this case than in the above experiment to achieve saturation of the first substrate layers by intercalated Li. Subsequently, a comparable submonolayer amount of [BMP][TFSA] was evaporated on the Li modified TiO2(110) substrate. XP spectra recorded after IL deposition and subsequent heating steps are shown in Fig. 9. Already after IL deposition at 300 K decomposition products are visible in the spectra (topmost spectra), in contrast to deposition on pure TiO2(110). Apparently, the surface species formed during the preceding Li intercalation process, Ti3+ species and possibly also surface near Li+ species, partly react with the [BMP][TFSA]. Here it should be noted that Ti3+ surface species were reported to be more reactive than Ti4+ species already mentioned previously.63 Therefore, in the Ti2p regime the Ti3+ peak intensity decreases relative to the Ti4+ peak (see also our above discussion). Overall, the new peaks formed upon [BMP][TFSA] decomposition are largely identical to those obtained for the reaction of post-deposited Li with a pre-deposited [BMP][TFSA] adlayer. The total amount of decomposition products, however, is smaller in the present case. In particular the decomposition peaks in the S2p region are less intense, and the Nanion peak does not show a strong decay. The amount of LiF (=Fd) and LixN (=NLixN) species is comparable. These differences may be due to the lower availability of the Ti3+ species due to diffusion limitations, as compared to direct deposition of Li on predeposited [BMP][TFSA] layers.
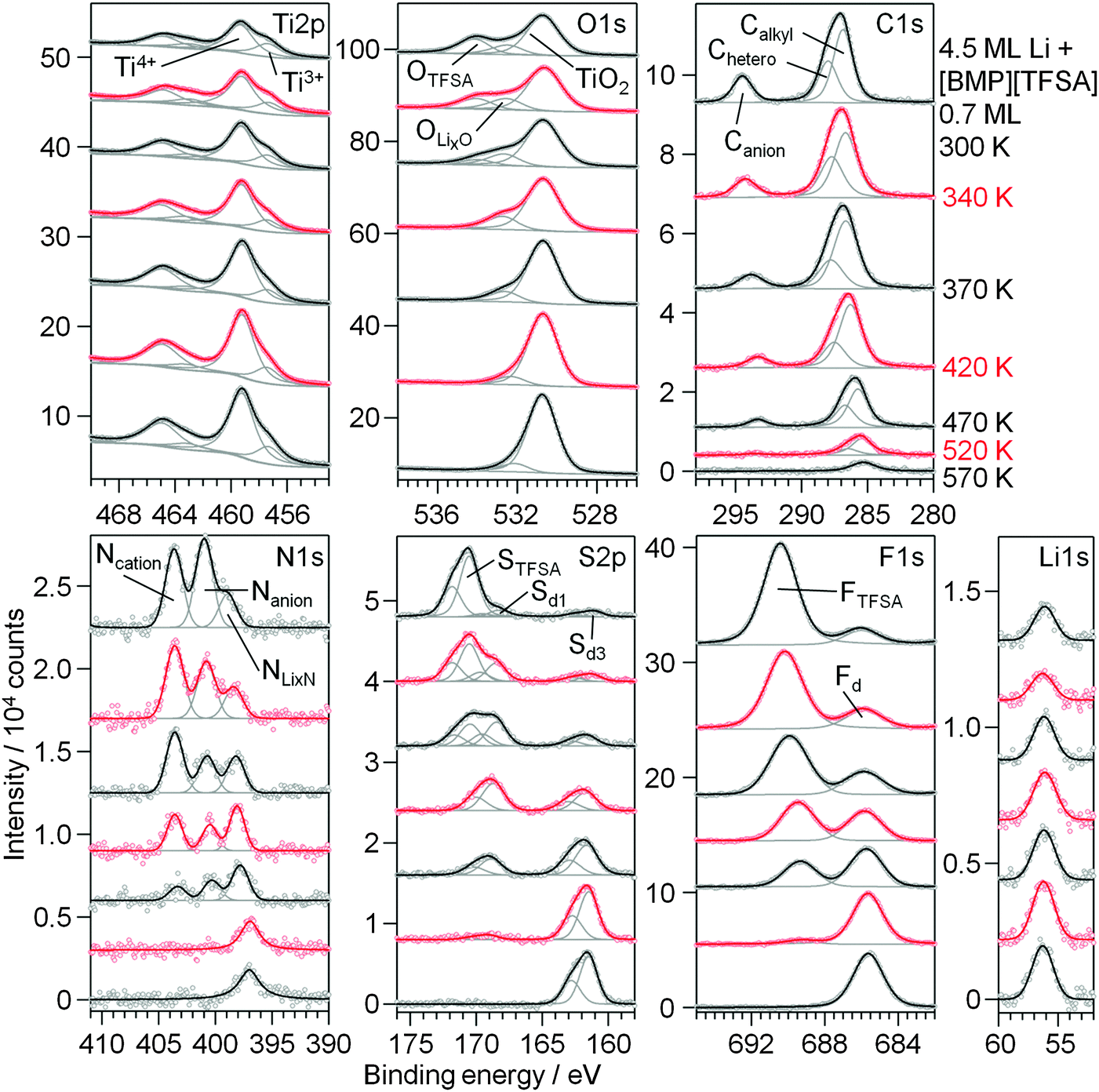 | ||
| Fig. 9 XP detail spectra of 0.7 ML [BMP][TFSA] post-deposited on a TiO2(110) surface pre-covered by 4.5 ML Li at 300 K (topmost spectra) and upon stepwise heating to 570 K. | ||
Upon sequential heating of the sample (lower spectra in Fig. 9), the OTFSA peak as well as the STFSA signal vanished at 370–420 K, as it was also the case for Li|IL|TiO2(110). In the same temperature range, Ti3+ again has a minimum in intensity. The peaks remaining after this heating step, the Canion (BE 293.3 eV), Nanion (BE 400.5 eV), Sd1 (BE 168.7 eV), OLixO (BE 532.8 eV) and FTFSA (BE 689.5 eV) peaks, are again related to the formation of F3C–O2S–N−Li+. They also vanished upon heating to 520 K, indicating the further decomposition of this product. The [BMP]+ cations are presumably also completely decomposed at 420 K and the remaining Calkyl, Chetero and Ncation peaks are due to remaining CxHyNz species, which decompose until 520 K. Above this temperature the surface mainly contains adsorbed LiF and Li2S (or/and Sad) species, and a small amount of remaining carbonaceous species are visible. Finally, a peak remaining in the N1s region with a BE of 397 eV, which at T > 520 K is shifted by 1 eV to lower BE compared to NLixN, is again attributed to TiNx species. For annealing to higher temperatures, e.g. to 630 K, we would expect this species to disappear as well, similar to our findings for annealing of a pure [BMP][TFSA] adlayer on TiO2(110).
Overall, the thermal decomposition of [BMP][TFSA] in interaction with codeposited Li does not seem to depend on the sequence of evaporation, whether Li is evaporated first followed by [BMP][TFSA] deposition or the other way around.
3 Summary and conclusion
Employing XPS and STM measurements for chemical and structural characterization, we have investigated the interaction of [BMP][TFSA] with a well-defined TiO2(110) surface, and the influence of Li thereon. For comparison, the interaction of Li with the bare TiO2(110) substrate was investigated as well. The results of the present study in combination with previous results lead us to the following conclusions:(1) In the temperature range between 80 K and 380 K, [BMP][TFSA] adsorbs as intact ion pairs on the substrate, with both anion and cation in direct contact with the surface. At 80 K, the [BMP][TFSA] ion pairs are adsorbed in lines along the TiO2(110) (1 × 1) [001] direction and uniformly cover the surface. The adsorbed species are highly mobile, molecular resolution STM imaging of the IL was only possible at this temperature and for a coverage of around 1 ML, while for other conditions only frizzy features could be resolved.
(2) Above 380 K, the adsorbed IL starts to decompose in a complex multistep mechanism. Sad, Fad, TiNx and also adsorbed CxHyN species are detected by XPS, which at T > 530 K further decompose to carbonaceous species. The reaction products fit to earlier proposals for the [BMP][TFSA] decomposition based on quantum chemical calculations.52–54 According to this mechanism the CxHyNz species are presumably [BMP]+ species with one of the C–N bonds broken, while the carbon containing species remaining at higher temperatures are butyl and methyl fragments, which can further decompose. The decomposition of the intact [BMP][TFSA] is completed after heating to 560 K.
(3) Li reacts already at 80 K with the TiO2(110) substrate, by intercalation into the near surface region of the TiO2(110) crystal, which leads to the formation of Li+ and Ti3+ species, and, at higher coverages, by the formation of a LixO cover layer. Intercalation results in a stoichiometry which is very close to that of Li loaded LTO (Li7Ti5O12). Upon heating to room temperature, the Ti3+ and Li+ species move deeper into the TiO2(110) bulk, but for the amounts investigated here (up to 9 ML equivalents of Li) they remain in the near surface region (in the topmost 5 nm), as evidenced by XPS measurements at the normal detection angle of the crystal.
(4) Deposition of Li at 80 K onto a TiO2(110) substrate pre-covered by a [BMP][TFSA] submonolayer film results in two competing reactions: reactive interaction of the Li atoms with the [BMP][TFSA] adlayer and intercalation into the TiO2(110) substrate forming intercalated Li+ and Ti3+ species. Reactive interaction between the IL adsorbate and Li results in decomposition products which are rather similar to those obtained for thermal decomposition of [BMP][TFSA] on TiO2(110), such as Li2S/Sad or LiF/Fad, but also additional peaks are found in XPS. Based on the XP binding energies, F3C–O2S–N−Li+ and F3C–O2S−Li+ species are formed as well as intermediate products such as SOx, in agreement with the theoretical proposals mentioned above. For Li deposition with pre-deposited multilayer [BMP][TFSA] films, the reaction with the IL is more efficient than diffusion of Li to the TiO2(110) surface, and reactive interaction with the TiO2(110) surface (intercalation) is therefore inhibited.
(5) For temperatures up to ca. 340 K, the adlayer composition changes only little. Mainly the Sd2 peak vanishes, indicating that F3C–O2S−Li+ is further decomposed. At higher temperatures further decomposition takes place: the remaining intact [BMP][TFSA] as well as the intermediate decomposition products F3C–O2S–N−Li+, SOx and CxHyNz species react with the Ti3+ and Li+ species in the substrate, forming similar reaction products as obtained for the thermal decomposition of [BMP][TFSA] on TiO2(110), such as Fad/LiF, Sad/Li2S and TiNx/LixN and some residual carbonaceous compounds. The decomposition of [BMP][TFSA] upon reactive interaction with TiO2(110) and Li is complete at a temperature of 420 K, the one of F3C–O2S–N−Li+ at 520–570 K. The Ti3+ species consumed in the decomposition process are reformed above 420 K. When evaporating [BMP][TFSA] on a TiO2(110) surface pre-covered with Li, the IL decomposes partly already at 300 K. Since this was not the case on pure TiO2(110), this higher reactivity is mainly attributed to the Ti3+ species intercalated in the near surface region. Upon heating to higher temperatures the decomposition process of [BMP][TFSA] is similar to that on samples where Li is deposited on TiO2(110) pre-covered with [BMP][TFSA].
Overall, this study gained detailed insight on a molecular level on the reactive interaction of [BMP][TFSA] with a TiO2(110) substrate and on the effect of Li on this interaction. These insights will be highly valuable and provide a solid basis for subsequent studies on the interaction between IL based electrolytes and TiO2 electrodes and on the initial stages of the formation of the solid-electrolyte interphase (SEI) between these compounds in lithium ion battery applications.
4 Experimental
The measurements were performed in a commercial UHV system (SPECS GmbH) with a base pressure of < 2 × 10−10 mbar. It is equipped with a combined Aarhus-type STM/AFM system (SPECS; Aarhus SPM 150 with Colibri Sensor), a non-monochromatic X-ray source (SPECS XR50, Al-Kα and Mg-Kα) and a hemispherical analyzer (SPECS, DLSEGD-Phoibos-HAS3500) for XPS measurements, and standard facilities for surface preparation. STM as well as XPS measurements can be carried out with the sample between 90–400 K by cooling with LN2 and resistive heating; for XPS measurements the sample can also be heated to temperatures up to 1000 K by electron bombardment (from the backside of the sample). All STM images were acquired in constant current mode with tunnelling currents between 20 and 200 pA and tunnelling voltages of 0.1–2 V. For XPS measurements we used an Al Kα X-ray source (1486.6 eV), operated at a power of 150 W (U = 12 kV, I = 12.5 mA). The spectra were recorded at a pass energy of 100 eV at detection angles of 0° and 80° relative to the surface normal. At 80° the surface sensitivity of the measurement is significantly higher than at normal emission (0° to the surface normal). The spectra shown in the figures are all taken at an electron emission angle of 80° if not stated otherwise. Peak fitting was performed with CasaXPS as well as Igor Pro 6. For background subtraction a Shirley background (if necessary convoluted with a parabolic background) was used, the peaks were fitted using a Pseudo-Voigt type peak shape, which is a convolution of a Gaussian and Lorentzian function. For the background subtraction of the Li peaks it was additionally necessary to subtract the signal of the empty TiO2 surface before performing the Shirley background subtraction due to the small intensity of the Li1s peak.Rutile TiO2(110) crystals were purchased from MaTeck GmbH with one side polished with an orientation accuracy of <0.1° and a surface roughness <0.03 μm. The initial preparation of the rutile TiO2 crystals follows procedures reported in the literature:76 the crystals were cleaned in an ultrasonic bath in 2× 5 min acetone, 5 min in caroic acid and 2× 5 min in ultrapure water (in between the steps the crystals were rinsed with ultrapure water). Afterwards the crystals were calcined at 1173 K under air atmosphere and cooled down at 5 K min−1. This procedure produces clean and fully oxidized colorless or slightly yellow rutile crystals. These crystals are transferred into the UHV chamber and heated in UHV to 1023 K for 5 h which partly reduces the sample (formation of Ti3+ species and oxygen vacancies, details can be found elsewhere18,46). Upon reduction, the color of the rutile crystal changes to blue. To achieve a properly prepared smooth (110) surface several cycles of Ar+ ion sputtering (0.5 kV, 45 min) and heating to 973 K for 30 min were carried out. The surface structure and cleanness were checked by STM and XPS (see e.g.Fig. 2a, b and 5) The surfaces show the typical (1 × 1) reconstruction reported by Diebold et al.17,18 with protruding rows running in the [001] direction. Bright round protrusions situated in between the bright rows are attributed to missing bridge-bonded oxygens on the surface. Terraces range over >100 nm. The XP spectra also show a contamination free substrate.
The ionic liquid [BMP][TFSA] was purchased from Merck in the highest available quality (ultrapure). It was filled in a glass crucible, which prior to use was baked at 870 K in UHV to remove vaporizable impurities from the glass. The crucible with the IL was positioned in a triple Knudsen effusion evaporator (ventiotec, OVD-3) and attached to the UHV chamber. The IL was degassed for several weeks under UHV conditions at room temperature and subsequently for at least 24 hours at 350 K which removes all vaporizable contaminations (such as water) from the IL. Karl-Fischer titration of the IL after room temperature degassing revealed water contents of well below 25 ppm, the water content after 350 K degassing should be considerably lower. Furthermore, the cleanness of the IL vapor was checked using a quadrupole mass spectrometer (QMS; Pfeiffer HiQuadQMG700), which was mounted with its ionization area into the IL beam; also the evaporation rate was calibrated against the evaporator temperature using the QMS. An evaporation temperature of 453 K and 5 min evaporation time results in a coverage of 1 ML. The [BMP][TFSA] adlayers were found to slowly decompose in the X-ray beam during XPS measurements, as it is already known from previous experiments applying non-monochromatized X-ray radiation.77 We quantified the beam damage occurring on our samples by measuring the decay of all peaks in the XP spectra assigned to [BMP][TFSA] over several hours. A decomposition rate of ca. 0.002 min−1 of the total intensities of the peaks for both the anion as well as the cation was found for a [BMP][TFSA] adlayer with a coverage of 0.7 ML at room temperature and with the X-ray source operated at 150 W. Decomposition products due to beam damage could not be detected in the spectra, which is clearly different to the decomposition due to addition of Li or the thermal decomposition of the adlayers described in Sections 2.1 and 2.3. We assume that the reaction products formed during exposure to X-ray radiation are desorbed right after formation. The decomposition mechanism under X-ray radiation is therefore completely different from the one induced thermally and/or Li incorporation. To minimize the effects of the beam damage in our measurements, the time for irradiation per sample was kept as short as possible, switching off the X-ray source between the XPS measurements. Furthermore, in the annealing experiments the heating was switched off before recording the XP spectra, these were therefore recorded at temperatures considerably lower than the respective annealing temperatures. In the evaluation of the XPS data the loss of intensity due to beam damage is taken into account, using the decomposition rate determined before.
Li was evaporated from a Li metal dispenser (SAES Getters) incorporated into a homemade electrical heating stage. The dispenser was operated at 7.1 A. The sample was placed in line-of-sight in front of the dispenser, the evaporation time was controlled with a shutter in between the sample and the dispenser. For calibration of the Li evaporation rate a Cu(111) crystal was used, which was cooled to 80 K. Under these conditions Li adsorbs atomically on the surface, it does not react with the substrate and only very slowly with residual contaminations adsorbing from the UHV gas phase.28 The coverage was evaluated by the damping of the Cu2p3/2 peak. For our setup a deposition rate of 0.049 ± 0.009 ML min−1 was calculated. The cleanness of the deposited Li adlayers both on Cu(111) and on TiO2(110) was checked by XPS and partly by STM.
Acknowledgements
This work was supported by the German Federal Ministry of Education and Research via the project “Li-Eco-Safe” under contract number 03X4636C within the program “Werkstoffinnovationen für Industrie und Gesellschaft”. B. U. is grateful for a fellowship by the Fonds der Chemischen Industrie. The authors would like to thank Dr J. Bansmann for advice in the evaluation of the XP spectra and Dorothea Alwast for help in the initial cleaning of the rutile TiO2(110) samples.References
- J. S. Wilkes, Green Chem., 2002, 4(2), 73–80 RSC.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39(21), 3772–3789 CrossRef CAS.
- G. B. Appetecchi, M. Montanino, D. Zane, M. Carewska, F. Alessandrini and S. Passerini, Electrochim. Acta, 2009, 54(4), 1325–1332 CrossRef CAS.
- J. M. Slattery, C. Daguenet, P. J. Dyson, T. J. S. Schubert and I. Krossing, Angew. Chem., 2007, 119(28), 5480–5484 CrossRef.
- M. Armand and J. M. Tarascon, Nature, 2008, 451(7179), 652–657 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8(8), 621–629 CrossRef CAS PubMed.
- A. A. Kornyshev, J. Phys. Chem. B, 2007, 111(20), 5545–5557 CrossRef CAS PubMed.
- F. Silva, C. Gomes, M. Figueiredo, R. Costa, A. Martins and C. M. Pereira, J. Electroanal. Chem., 2008, 622(2), 153–160 CrossRef CAS.
- B. Uhl, T. Cremer, M. Roos, F. Maier, H. P. Steinrück and R. J. Behm, Phys. Chem. Chem. Phys., 2013, 15, 17295–17308 RSC.
- F. Buchner, K. Forster-Tonigold, B. Uhl, D. Alwast, N. Wagner, H. Farkhondeh, A. Groß and R. J. Behm, ACS Nano, 2013, 7(9), 7773–7784 CrossRef CAS PubMed.
- B. Uhl, F. Buchner, D. Alwast, N. Wagner and R. J. Behm, Beilstein J. Nanotechnol., 2013, 4(1), 903–918 CrossRef CAS PubMed.
- B. Uhl, F. Buchner, S. Gabler, M. Bozorgchenani and R. J. Behm, Chem. Commun., 2014, 50(62), 8601–8604 RSC.
- B. Uhl, H. Huang, D. Alwast, F. Buchner and R. J. Behm, Phys. Chem. Chem. Phys., 2015, 17, 23816–23832 RSC.
- Z. Chen, I. Belharouak, Y.-K. Sun and K. Amine, Adv. Funct. Mater., 2013, 23(8), 959–969 CrossRef CAS.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- T. Fröschl, U. Hörmann, P. Kubiak, G. Kucerova, M. Pfanzelt, C. K. Weiss, R. J. Behm, N. Hüsing, U. Kaiser and K. Landfester, Chem. Soc. Rev., 2012, 41(15), 5313–5360 RSC.
- U. Diebold, Appl. Phys. A: Mater. Sci. Process., 2003, 76(5), 681–687 CrossRef CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48(5), 53–229 CrossRef CAS.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115(13), 6357–6426 CrossRef CAS PubMed.
- S. Krischok, J. A. Schaefer, O. Höfft and V. Kempter, Surf. Interface Anal., 2004, 37(1), 83–89 CrossRef.
- H. Tatsumi, A. Sasahara and M. Tomitori, J. Phys. Chem. C, 2012, 116(25), 13688–13692 CAS.
- A. Henningsson, M. P. Andersson, P. Uvdal, H. Siegbahn and A. Sandell, Chem. Phys. Lett., 2002, 360(1), 85–90 CrossRef CAS.
- A. Henningsson, H. Rensmo, A. Sandell, H. Siegbahn, S. Södergren, H. Lindström and A. Hagfeldt, J. Chem. Phys., 2003, 118, 5607–5612 CrossRef CAS.
- S. Södergren, H. Siegbahn, H. Rensmo, H. Lindström, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1997, 101(16), 3087–3090 CrossRef.
- M. Pfanzelt, Development of TiO2 Rutile as Negative Electrode Material for Lithium Ion Batteries, PhD thesis, University Ulm, ZSW, 2012 Search PubMed.
- B. Zachau-Christiansen, K. West, T. Jacobsen and S. Atlung, Solid State Ionics, 1988, 28, 1176–1182 CrossRef.
- M. Olschewski, R. Gustus, M. Marschewski, O. Höfft and F. Endres, Phys. Chem. Chem. Phys., 2014, 16(47), 25969–25977 RSC.
- F. Buchner, M. Bozorgchenani, B. Uhl, H. Farkhondeh, J. Bansmann and R. J. Behm, J. Phys. Chem. C, 2015, 119(29), 16649–16659 CAS.
- U. Diebold and T. E. Madey, Surf. Sci. Spectra, 1996, 4(3), 227–231 CrossRef CAS.
- T. Cremer, M. Killian, J. M. Gottfried, N. Paape, P. Wasserscheid, F. Maier and H. P. Steinrück, ChemPhysChem, 2008, 9(15), 2185–2190 CrossRef CAS PubMed.
- T. Cremer, M. Stark, A. Deyko, H. P. Steinrück and F. Maier, Langmuir, 2011, 27, 3662–3671 CrossRef CAS PubMed.
- T. Cremer, L. Wibmer, S. K. Calderon, A. Deyko, F. Maier and H. P. Steinrück, Phys. Chem. Chem. Phys., 2012, 14(15), 5153–5163 RSC.
- U. Diebold, J. Lehman, T. Mahmoud, M. Kuhn, G. Leonardelli, W. Hebenstreit, M. Schmid and P. Varga, Surf. Sci., 1998, 411(1), 137–153 CrossRef CAS.
- S. Fischer, A. W. Munz, K. D. Schierbaum and W. Göpel, Surf. Sci., 1995, 337(1), 17–30 CrossRef CAS.
- K. i. Fukui, H. Onishi and Y. Iwasawa, Phys. Rev. Lett., 1997, 79(21), 4202–4205 CrossRef CAS.
- S. Wendt, R. Schaub, J. Matthiesen, E. K. Vestergaard, E. Wahlström, M. D. Rasmussen, P. Thostrup, L. M. Molina, E. Laegsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Surf. Sci., 2005, 598, 226–245 CrossRef CAS.
- Z. Zhang, J. Lee, J. T. Yates, R. Bechstein, E. Lira, J. Hansen, S. Wendt and F. Besenbacher, J. Phys. Chem. C, 2010, 114(7), 3059–3062 CAS.
- E. Lira, J. Hansen, P. Huo, R. Bechstein, P. Galliker, E. Laegsgaard, B. Hammer, S. Wendt and F. Besenbacher, Surf. Sci., 2010, 604(21), 1945–1960 CrossRef CAS.
- J. Hansen, P. Huo, U. Martinez, E. Lira, Y. Y. Wei, R. Streber, E. Laegsgaard, B. Hammer, S. Wendt and F. Besenbacher, Phys. Rev. Lett., 2011, 107(13), 136102 CrossRef CAS PubMed.
- U. Martinez, J. Hansen, E. Lira, H. H. Kristoffersen, P. Huo, R. Bechstein, E. Laegsgaard, F. Besenbacher, B. Hammer and S. Wendt, Phys. Rev. Lett., 2012, 109(15), 155501 CrossRef CAS PubMed.
- R. Wen, B. Rahn and O. M. Magnussen, Angew. Chem., 2015, 127(20), 5889 CrossRef.
- T. Waldmann, H. H. Huang, H. E. Hoster, O. Höfft, F. Endres and R. J. Behm, ChemPhysChem, 2011, 12, 2565–2567 CrossRef CAS PubMed.
- B. J. Burrow, A. E. Morgan and R. C. Ellwanger, J. Vac. Sci. Technol., A, 1986, 4(6), 2463–2469 CAS.
- J. A. Rodriguez, J. Hrbek, J. Dvorak, T. Jirsak and A. Maiti, Chem. Phys. Lett., 2001, 336(5), 377–384 CrossRef CAS.
- E. L. D. Hebenstreit, W. Hebenstreit and U. Diebold, Surf. Sci., 2001, 470(3), 347–360 CrossRef CAS.
- E. L. D. Hebenstreit, W. Hebenstreit and U. Diebold, Surf. Sci., 2000, 461(1), 87–97 CrossRef CAS.
- A. M. Czoska, S. Livraghi, M. Chiesa, E. Giamello, S. Agnoli, G. Granozzi, E. Finazzi, C. D. Valentin and G. Pacchioni, J. Phys. Chem. C, 2008, 112(24), 8951–8956 CAS.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 14(9), 3808–3816 CrossRef CAS.
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108(13), 4086–4093 CrossRef CAS.
- R. E. Banks, B. E. Smart and J. C. Tatlow, Organofluorine Chemistry: Principles and Commercial Applications, Springer, 1994 Search PubMed.
- S. Schernich, M. Laurin, Y. Lykhach, H. P. Steinrück, N. Tsud, T. Skala, K. C. Prince, N. Taccardi, V. Matolin, P. Wasserscheid and J. Libuda, J. Phys. Chem. Lett., 2013, 4(1), 30–35 CrossRef CAS PubMed.
- M. C. Kroon, W. Buijs, C. J. Peters and G. J. Witkamp, Green Chem., 2006, 8(3), 241–245 RSC.
- P. C. Howlett, E. I. Izgorodina, M. Forsyth and D. R. MacFarlane, Z. Phys. Chem., 2006, 220(10), 1483–1498 CrossRef CAS.
- M. C. Kroon, W. Buijs, C. J. Peters and G. J. Witkamp, Thermochim. Acta, 2007, 465(1), 40–47 CrossRef CAS.
- R. E. Del Sesto, T. M. McCleskey, C. Macomber, K. C. Ott, A. T. Koppisch, G. A. Baker and A. K. Burrell, Thermochim. Acta, 2009, 491(1), 118–120 CrossRef CAS.
- S. E. Stein, “Mass Spectra” by NIST Mass Spec Data Center, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899, http://webbook.nist.gov, 2015.
- C. Xu, B. E. Koel and M. T. Paffett, Langmuir, 1994, 10(1), 166–171 CrossRef CAS.
- S. L. Tait, Z. Dohnalek, C. T. Campbell and B. D. Kay, J. Chem. Phys., 2005, 122(16), 164707 CrossRef PubMed.
- R. M. Slayton, C. M. Aubuchon, T. L. Camis, A. R. Noble and N. J. Tro, J. Phys. Chem., 1995, 99(7), 2151–2154 CrossRef CAS.
- P. E. Pehrsson, P. Armistead and S. Sastri, Surf. Interface Anal., 1993, 20(7), 573–582 CrossRef CAS.
- K. Hamrin, G. Johansson, U. Gelius, C. Nordling and K. Siegbahn, Phys. Scr., 1970, 1(5–6), 277–280 CrossRef CAS.
- D. R. Warburton, D. Purdie, C. A. Muryn, K. Prabhakaran, P. L. Wincott and G. Thornton, Surf. Sci., 1992, 269, 305–309 CrossRef.
- K. E. Smith, J. L. Mackay and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35(11), 5822–5829 CrossRef CAS.
- H. Onishi, T. Aruga, C. Egawa and Y. Iwasawa, Surf. Sci., 1988, 193(1), 33–46 CrossRef CAS.
- K. E. Smith and V. E. Henrich, J. Vac. Sci. Technol., A, 1989, 7(3), 1967–1971 CAS.
- S. P. Kowalczyk, L. Ley, F. R. McFeely, R. A. Pollak and D. A. Shirley, Phys. Rev. B: Solid State, 1973, 8(8), 3583–3585 CrossRef CAS.
- L. El Ouatani, R. Dedryvere, C. Siret, P. Biensan and D. Gonbeau, J. Electrochem. Soc., 2009, 156(6), A468–A477 CrossRef CAS.
- L. Liu, V. E. Henrich, W. P. Ellis and I. Shindo, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(3), 2236–2239 CrossRef CAS.
- A. C. Kozen, A. J. Pearse, C. F. Lin, M. A. Schroeder, M. Noked, S. B. Lee and G. W. Rubloff, J. Phys. Chem. C, 2014, 118(48), 27749–27753 CAS.
- M. P. Seah and S. J. Spencer, Surf. Interface Anal., 2002, 33(8), 640–652 CrossRef CAS.
- M. Kitta, T. Akita, Y. Maeda and M. Kohyama, Langmuir, 2012, 28(33), 12384–12392 CrossRef CAS PubMed.
- B. Lindberg, K. Hamrin, G. Johansson, U. Gelius, A. Fahlman, C. Nordling and K. Siegbahn, Phys. Scr., 1970, 1(5–6), 286–298 CrossRef CAS.
- K. R. Zavadil and N. R. Armstrong, Surf. Sci., 1990, 230(1), 61–73 CrossRef CAS.
- X. Feng, M. K. Song, W. C. Stolte, D. Gardenghi, D. Zhang, X. Sun, J. Zhu, E. J. Cairns and J. Guo, Phys. Chem. Chem. Phys., 2014, 16(32), 16931–16940 RSC.
- C. Xu, B. Sun, T. Gustafsson, K. Edström, D. Brandell and M. Hahlin, J. Mater. Chem. A, 2014, 2(20), 7256–7264 CAS.
- S. Kielbassa, M. Kinne and R. J. Behm, J. Phys. Chem. B, 2004, 108(50), 19184–19190 CrossRef CAS.
- A. Keppler, M. Himmerlich, T. Ikari, M. Marschewski, E. Pachomow, O. Höfft, W. Maus-Friedrichs, F. Endres and S. Krischok, Phys. Chem. Chem. Phys., 2011, 13, 1174–1181 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07433a |
| This journal is © the Owner Societies 2016 |