
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGrasping hydrogen adsorption and dynamics in metal–organic frameworks using 2H solid-state NMR†
Bryan E. G.
Lucier‡
,
Yue
Zhang‡
,
Kelly J.
Lee
,
Yuanjun
Lu
and
Yining
Huang
*
Department of Chemistry, The University of Western Ontario, 1151 Richmond Street, London, Ontario N6A 5B7, Canada. E-mail: yhuang@uwo.ca
First published on 4th May 2016
Abstract
Record greenhouse gas emissions have spurred the search for clean energy sources such as hydrogen (H2) fuel cells. Metal–organic frameworks (MOFs) are promising H2 adsorption and storage media, but knowledge of H2 dynamics and adsorption strengths in these materials is lacking. Variable-temperature (VT) 2H solid-state NMR (SSNMR) experiments targeting 2H2 gas (i.e., D2) shed light on D2 adsorption and dynamics within six representative MOFs: UiO-66, M-MOF-74 (M = Zn, Mg, Ni), and α-M3(COOH)6 (M = Mg, Zn). D2 binding is relatively strong in Mg-MOF-74, Ni-MOF-74, α-Mg3(COOH)6, and α-Zn3(COOH)6, giving rise to broad 2H SSNMR powder patterns. In contrast, D2 adsorption is weaker in UiO-66 and Zn-MOF-74, as evidenced by the narrow 2H resonances that correspond to rapid reorientation of the D2 molecules. Employing 2H SSNMR experiments in this fashion holds great promise for the correlation of MOF structural features and functional groups/metal centers to H2 dynamics and host–guest interactions.
Carbon dioxide (CO2) is associated with the greenhouse effect and global warming. H2 fuel cells and other “greener” solutions are the future energy sources for automobiles, however, many proposals for H2 storage involve tanks for compressed H2,1 which is flammable and explosive. Safer alternatives for H2 storage, such as crystalline materials,1,2 are desired. Metal–organic frameworks (MOFs) are ordered three-dimensional structures consisting of metal centers or metal–inorganic units joined by organic linkers. By varying the MOF topology, metal center, and linkers,3 tailored large surface areas and guest binding strengths are possible. Several MOFs have shown H2 adsorption and storage capabilities,4 including UiO-66,5,6 MOF-74,7 α-Mg3(COOH)6,8 and α-Zn3(COOH)6.9
X-ray diffraction is widely used to investigate gas adsorption in MOFs but cannot reliably locate H2. Neutron diffraction can find H2 but little motional details can be obtained. Adsorption isotherms provide H2 capacity yet yield little positional or dynamic data. IR spectroscopy may indicate H2 adsorption and binding sites, but H2 motion remains unknown. Computational methods can estimate H2 location and dynamics, yet demand experimental verification. In order to (i) move MOFs toward practical incorporation as H2 storage media, and (ii) enhance H2 capacity in future MOFs, knowledge of the dynamic behavior of H2 in today's MOFs is critical. SSNMR is a sensitive probe of the local nuclear electronic and magnetic environment, and provides rich information on MOFs from the perspective of the metals,10 organic linkers,11 guest molecules,12 and dynamic components.13,14 NMR interactions are generally anisotropic (directionally-dependent) with respect to the magnetic field and are influenced by dynamics in a predictable manner; rich motional information can be extracted from spectral simulations.15
When dynamics are of interest in NMR, the 2H isotope is preferred over 1H. 2H has a spin of 1 and is subject to the anisotropic quadrupolar interaction (QI) between the nuclear quadrupole moment and surrounding electric field gradients (EFGs). Any motion that reorients the 2H EFG tensor influences the QI; since the 2H SSNMR spectrum is dominated by the QI, 2H SSNMR is a powerful probe of guest dynamics. 1H is a spin-1/2 nucleus that suffers from very strong 1H–1H homonuclear dipolar coupling in solids, resulting in broad 1H SSNMR spectra that rarely yield useful information. 2H SSNMR has proven to be an effective tool for studying the binding in metal–dihydrogen complexes,16–18 and has been successfully used to probe D2 mobility within a Ru-modified MOF.19 Wright et al. have shown that 2H SSNMR provides rich information on the dynamics of deuterated linkers and guests in MOFs and microporous materials.20–24 Herein, we use VT 2H SSNMR to probe H2 adsorption in a series of different MOFs, studying the significant differences in D2 adsorption behavior and dynamics within UiO-66, M-MOF-74 (M = Zn, Mg, Ni), and α-M3(COOH)6 (M = Mg, Zn).
UiO-666 is a three-dimensional MOF composed of Zr6O4(OH)4 units and 1,4-benzenedicarboxylate (BDC) linkers, with large octahedral and tetrahedral cages with pore sizes ca. 11 Å and 8 Å, respectively (Fig. S1, ESI†). VT 2H SSNMR spectra of D2 in UiO-66 (Fig. S2, ESI†) feature a narrow resonance from 293 K to 133 K with a full width at half height (FWHH) of 45 Hz throughout. Although D2 is adsorbed in UiO-66, the large pores permit rapid diffusion of D2 guests through the MOF void space,5 eliminating the spectral broadening effects of the QI and giving rise to a sharp, motionally-averaged 2H resonance. The narrow lineshape indicates that any localized MOF–D2 interactions are relatively weak and easily overcome by diffusion. The tetrahedral and octahedral pore geometries may also contribute to the observed narrow resonance.
M-MOF-74 (M = metal) is composed of metal centers connected by 2,5-dioxido-1,4-benzenedicarboxylate (dobdc) linkers that form honeycomb-shaped channels ca. 11 Å wide with metal centers at each vertex (Fig. S3, ESI†). Each metal center is connected to five oxygen atoms from four linkers in the as-made MOF, along with a sixth oxygen from a water molecule that can be removed via activation. The resulting coordinatively-unsaturated open metal site (OMS) strongly encourages adsorption of H2.7,25 The nature of the metal center influences H2 binding affinity, which increases in the order Zn < Mg < Ni.7 The 2H SSNMR spectrum of D2 in Zn-MOF-74 features a narrow resonance of 80 Hz FWHH at 293 K (Fig. 1(a)), implying that D2 undergoes rapid isotropic reorientation in the MOF. The FWHH increases to 120 Hz at 213 K and 1920 Hz at 133 K. At lower temperatures, D2 motion is reduced, leading to a large increase in FWHH. The 2H resonance widths in Zn-MOF-74 are much larger than in UiO-66 at all temperatures, indicating that D2 is considerably less mobile in Zn-MOF-74 due to metal–H2 interactions; however, the lack of spectral features at 133 K confirms that D2 dynamics remain rapid, and efficient H2 diffusion pathways in the MOF-74 family are known to exist.26
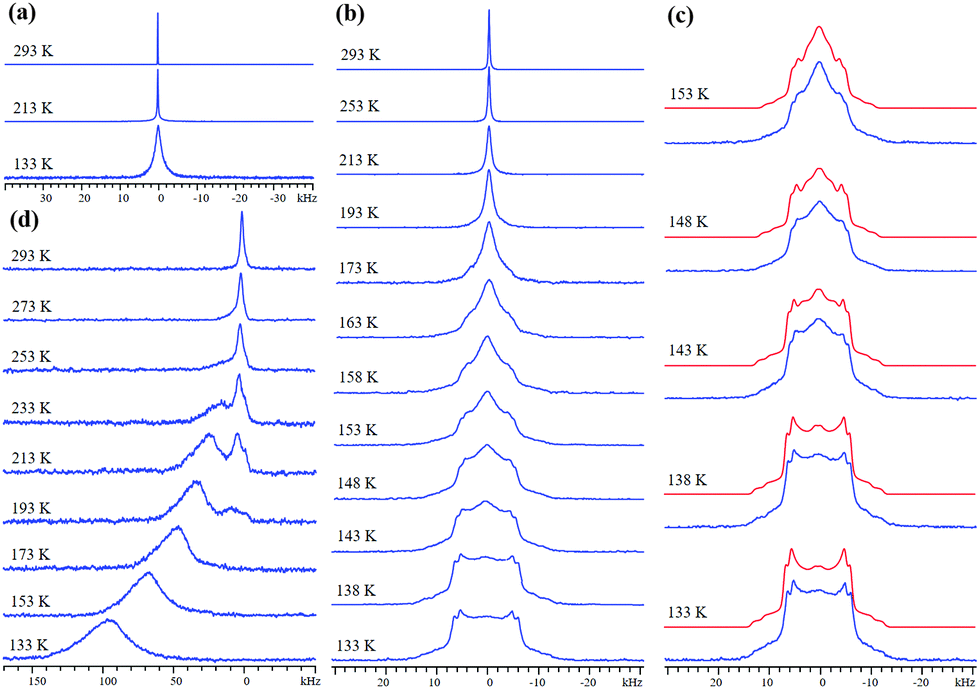 | ||
| Fig. 1 The experimental static VT 2H SSNMR spectra of adsorbed D2 within (a) Zn-MOF-74, (b and c) Mg-MOF-74, and (d) Ni-MOF-74 are depicted. Experimental (blue) and simulated (red) 2H SSNMR spectra in Mg-MOF-74 at low temperatures are shown in (c). The sharp resonance at ca. 0 kHz in all spectra at high temperatures corresponds to mobile or very weakly adsorbed D2; this resonance is broadened in (d) due to the influence of the paramagnetic Ni metal center. | ||
Mg-MOF-74 has a higher affinity for H2/D2 and yields the VT SSNMR spectra in Fig. 1(b). The narrow resonance at 293 K corresponds to highly mobile D2 gas, while emerging broad spectral features at 173 K indicate the onset of significant D2 adsorption. At 153 K, adsorbed D2 gives rise to two broad 2H powder patterns along with a narrow central resonance from free D2 in the center of the pores. The wide 2H lineshapes at 133 K are intense, well-defined, and associated with most of the D2 in Mg-MOF-74 (Fig. 1(c)). By comparing the 2H quadrupolar coupling constant (CQ) of gaseous D2 (225 kHz27) to the apparent CQs of D2 in Mg-MOF-74 (Table S1, ESI†), D2 dynamic information can be extracted.18 Observed CQ(2H) values in Mg-MOF-74 are far less than 225 kHz, and are also smaller than the CQ(2H) range of ca. 30–120 kHz associated with η2 bonding between a metal and D2 in metal–dihydrogen complexes,18,28–30 implying that fast D2 motion exists here and that the metal–D2 interactions in Mg-MOF-74 are weaker than the bonding in metal–dihydrogen complexes. It should be noted that at the experimental temperatures and magnitudes of quadrupolar coupling in this study, any spectral effects arising from D2 rotational tunneling are expected to be negligible.31,32
The same two powder patterns and NMR parameters persist, along with the sharp component, when the loading level is halved to 0.1 D2/Mg (Fig. S4 and S5, Table S2, ESI†). The two broad 2H powder patterns of D2 in Mg-MOF-74 reveal that there are two similar, but nonequivalent D2 molecules (D2(1) and D2(2)) adsorbed on two different OMSs, as indicated by their QI parameters and D2 motions (see Appendix A in the ESI†), while the narrow third resonance indicates that highly mobile D2 is also present. Previous studies have suggested that there may be two very similar H2 adsorption sites of nearly identical adsorption enthalpy localized on the OMS near our experimental loading levels with slightly different interaction geometries.33,34 There are 6 OMSs located near the same cross-sectional plane per channel (Fig. 2(b)). At a low loading level of 0.1 D2/metal, on average, there is less than one D2 guest per channel cross-section. Thus, it is reasonable to assume that no two D2 molecules are adsorbed simultaneously on the same OMS at any given time. It is likely that each channel cross-section contains only 1 D2 molecule that is in rapid exchange among the 6 sites (vide infra). Based on the intensity ratios in Table S1 (ESI†), ca. 59% of the channel cross-sections are populated by D2(1) and 41% of the channel cross-sections are occupied by D2(2).
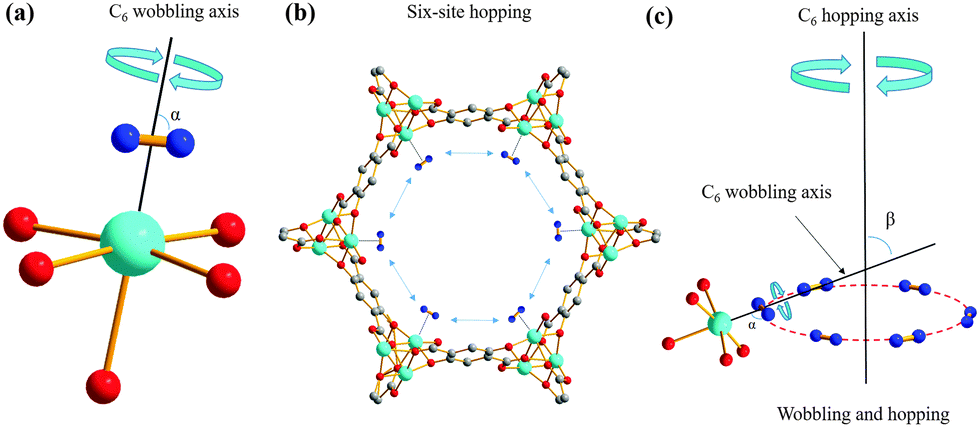 | ||
| Fig. 2 The localized C6 rotation, or wobbling, of D2 molecules adsorbed on the OMS is shown in (a). The black line represents the metal–D2 vector; the cone of D2 wobbling movement traces out an angle α about the wobbling axis. A schematic of D2 hopping in MOF-74 is shown in (b). The combination of wobbling (C6 rotation, α) and six-site hopping (C6 rotation, β) of D2 molecules in MOF-74 is shown in (c), where the wobbling axis makes an angle β with respect to the hopping axis. The colors red, grey, blue and cyan correspond to oxygen, carbon, deuterium and the metal center, respectively. Our data indicate that there are two very similar D2 adsorption sites on the OMS, giving rise to two separate 2H SSNMR powder patterns. | ||
Using the known 2H QI parameters of D2 gas (CQ(2H) = 225 kHz, ηQ = 0),27 simulations15 of motionally-averaged 2H spectra (Fig. S6, ESI†) reveal that common D2 dynamics exist at the two similar adsorption sites on the OMS in Mg-MOF-74 (Fig. 2 and Table S3, ESI†). D2 undergoes a local rotation or “wobbling” modeled by a sixfold rotation about the minimum energy configuration with respect to the OMS (Fig. 2(a)), as well as a non-localized six-site hopping along the pore edge (Fig. 2(b)). The combined motional model is shown in Fig. 2(c). The wobbling describes a rotation of the D–D bond about the axis passing through the OMS (Fig. 2(a)), as defined by angle α. It is likely that the wobbling rotation represents a model for some kind of rotational diffusion on the OMS. The hopping occurs between six nearly coplanar OMSs, where three reside in one plane, and the other three OMSs are in a very proximate plane slightly offset along the longitudinal direction of the channel. It should be noted that since the wobbling and hopping motional rates are in the fast exchange limit, there are an infinite number of Cn (n ≥ 3) jumping motions that could lead to the observed powder patterns. The dynamic behavior of D2 at both adsorption sites is strikingly similar, supporting the notion of two H2 adsorption sites of nearly identical interaction geometry and enthalpy on the OMS: each has D2 wobbling angles increasing from ca. 74° at 293 K to 82° at 133 K, along with constant hopping angles of ca. 60° (Table S3, ESI†). The larger wobbling angle at low temperatures enhances the interaction of D2 with the Mg site. It should also be noted that simulation of fast-exchange SSNMR spectra can sometimes lead to more than one motional model. However, we were unable to simulate the split “horns” or characteristic “shoulders” of the low-temperature spectra using a single adsorption site with any type of possible motions (Appendix A). These results also suggest that there does not seem to be significant exchange of D2 between the two adsorption sites; increasing the possibility that the sites are somehow segregated or correspond to slightly different types of OMSs in separate MOF channels. The calculated H2 position in Co-MOF-7435 strongly agrees with the orientations in our motional model, while the types of D2 dynamics resemble those of CO2 and CO in MOF-74.36–38 To confirm at low temperatures that the D2 motional rate remains in the fast (i.e., ≥107 Hz) and not in the intermediate regime, quadrupolar echo experiments with different inter-pulse delays were performed, resulting in unchanged spectra (Fig. S7 and S8, Table S4, ESI†).
Ni-MOF-74 has a stronger H2/D2 binding affinity.7 Unlike diamagnetic Zn2+ and Mg2+, Ni2+ is paramagnetic in this MOF. The 2H magnetic dipole couples with those of proximate unpaired electrons, resulting in spectral broadening and unusual chemical shifts when D2 is near Ni2+. VT 2H SSNMR spectra of D2 in Ni-MOF-74 (Fig. 2(d)) at room temperature reveal that D2 is mobile and/or rapidly exchanges with adsorbed D2. The broad 1.2 kHz FWHH confirms the proximity of D2 to Ni2+. At 273 K, a second, broader resonance emerges at 140 ppm, corresponding to D2 adsorbed on the OMS. At 213 K, the broad resonance is at 375 ppm and is the dominant feature. The broad resonance increases in frequency, width, and relative intensity as temperature is reduced; the narrower resonance at lower frequency vanishes at 173 K, implying that most D2 is adsorbed. At 133 K a broad, featureless 2H resonance of FWHH 30 kHz centered at about 1550 ppm is evident, confirming that a majority of D2 is adsorbed onto the OMS. The featureless nature of these resonances prohibits detailed analysis.39
α-Mg3(COOH)6 and α-Zn3(COOH)6 are porous MOFs8,9,40 with one-dimensional zig-zag channels of small ca. 4–5 Å diameter (Fig. S9, ESI†). The 2H spectrum of D2 in α-Mg3(COOH)6 at 293 K features a narrow resonance of 375 Hz FWHH (Fig. S10(a), ESI†) arising from mobile D2, flanked by a less intense powder pattern 40 kHz broad at 193 K from adsorbed D2, in agreement with adsorption isotherms.8,40 At 133 K the broad powder patterns are of higher S/N, reflecting increased D2 adsorption in α-Mg3(COOH)6, and are convoluted with a broad distribution of intensity possibly from disordered D2; other small guests such as ethanol and methanol are also disordered in this MOF.40 The 2H SSNMR spectra of D2 in α-Zn3(COOH)6 are of similar lineshape and breadth (Fig. S10(b), ESI†), implying that both MOFs have similar H2 adsorption strengths. The intense narrow resonance persists at lower temperatures in α-Zn3(COOH)6, indicating that there is more mobile H2 in α-Zn3(COOH)6 at all temperatures. The complicated powder patterns are clear evidence of multiple D2 adsorption sites within both α-Mg3(COOH)6 and α-Zn3(COOH)6. At 0.1 D2/metal loading, in addition to free gas, 2H spectra may be simulated using three and two adsorption sites in α-Mg3(COOH)6 and α-Zn3(COOH)6, respectively. In strong agreement with our experimental observations is a very recent study that has located three main H2 adsorption sites in α-Mg3(COOH)6.41
The 2H spectrum of α-Zn3(COOH)6 is less ambiguous, of higher resolution, and is more straightforward to simulate (Fig. S10(c), Tables S3 and S5, ESI†). The well-defined 2H SSNMR spectra of D2 in α-Zn3(COOH)6 yield relatively high ηQ values along with CQ values ca. 5–10 kHz greater than those of D2 in Mg-MOF-74 at all equivalent temperatures, reflecting reduced D2 mobility and perhaps a stronger D2-MOF interaction in α-Zn3(COOH)6. In contrast, the very complicated 2H spectra of α-Mg3(COOH)6 permit only a preliminary motional simulation at this time (Fig. S11, ESI†). A combined wobbling and hopping of D2 occurs at all adsorption sites in both systems. D2 hops between two adsorption sites in α-Zn3(COOH)6, reminiscent of the twofold hopping by CO2 guests in the α-M3(COOH)6 MOF.42 It is notable that the changes in D2 motional angles within Zn are not very pronounced across the experimental temperature range (Table S3, ESI†). At this point, the nature of the adsorption sites is unclear from SSNMR experiments. We are currently performing more detailed studies in order to extract detailed D2 motional and adsorption information from both α-M3(COOH)6 MOFs.
2H SSNMR spectroscopy has revealed unique insights into H2 dynamics and adsorption locations within these six MOFs; this information is unavailable or very difficult to obtain using traditional MOF characterization methods such as adsorption isotherms and X-ray diffraction. Complementary methods such as neutron diffraction, inelastic neutron scattering, and VT infrared spectroscopy are necessary to locate adsorption locations, but yield limited motional data. Variation of the MOF topology, metal center, and linker gives rise to very different D2 dynamic behaviors and adsorption strengths. Further studies employing lower temperatures (i.e., <100 K) are necessary to reduce H2 mobility and obtain richer dynamic knowledge in these systems, however, these experiments are not yet possible with the equipment available to us. We have used relatively low levels of D2 loading to simplify these systems for spectral simulations; the next step is to explore higher D2 loading levels in order to understand the implications for H2 motion in a practical hydrogen storage setting. The comprehensive molecular-level knowledge of H2 dynamics and adsorption in MOFs available from 2H SSNMR spectroscopy will undoubtedly assist in establishing clear links between H2 dynamics and high-capacity H2 storage in porous materials, with clear applications in green energy solutions.
Y. H. thanks the Natural Science and Engineering Research Council (NSERC) of Canada for a Discovery grant and a Discovery Accelerator Supplements Award. We also thank an anonymous Reviewer for the helpful input and suggestions.
References
- D. J. Durbin and C. Malardier-Jugroot, Int. J. Hydrogen Energy, 2013, 38, 14595–14617 CrossRef CAS.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- M. D. Allendorf and V. Stavila, CrystEngComm, 2015, 17, 229–246 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- W. Zhou, H. Wu and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 15268–15269 CrossRef CAS PubMed.
- B. Schmitz, I. Krkljus, E. Leung, H. W. Höffken, U. Müller and M. Hirscher, ChemSusChem, 2010, 3, 758–761 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, M. Kurmoo, T. Liu, S. Vilminot, B. Zhao and S. Gao, Aust. J. Chem., 2006, 59, 617–628 CrossRef CAS.
- P. He, B. E. G. Lucier, V. V. Terskikh, Q. Shi, J. X. Dong, Y. Y. Chu, A. M. Zheng, A. Sutrisno and Y. N. Huang, J. Phys. Chem. C, 2014, 118, 23728–23744 CAS.
- D. I. Kolokolov, A. G. Stepanov, V. Guillerm, C. Serre, B. Frick and H. Jobic, J. Phys. Chem. C, 2012, 116, 12131–12136 CAS.
- D. I. Kolokolov, H. Jobic, S. Rives, P. G. Yot, J. Ollivier, P. Trens, A. G. Stepanov and G. Maurin, J. Phys. Chem. C, 2015, 119, 8217–8225 CAS.
- K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514–519 CrossRef CAS PubMed.
- V. N. Vukotic, C. A. O'Keefe, K. Zhu, K. J. Harris, C. To, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2015, 137, 9643–9651 CrossRef CAS PubMed.
- R. L. Vold and G. L. Hoatson, J. Magn. Reson., 2009, 198, 57–72 CrossRef CAS PubMed.
- T. Gutmann, I. del Rosal, B. Chaudret, R. Poteau, H. H. Limbach and G. Buntkowsky, ChemPhysChem, 2013, 14, 3026–3033 CrossRef CAS PubMed.
- R. H. Morris, Coord. Chem. Rev., 2008, 252, 2381–2394 CrossRef CAS.
- G. A. Facey, T. P. Fong, D. Gusev, P. M. Macdonald, R. H. Morris, M. Schlaf and W. Xu, Can. J. Chem., 1999, 77, 1899–1910 CrossRef CAS.
- F. Schröder, D. Esken, M. Cokoja, M. W. E. van den Berg, O. I. Lebedev, G. Van Tendeloo, B. Walaszek, G. Buntkowsky, H.-H. Limbach, B. Chaudret and R. A. Fischer, J. Am. Chem. Soc., 2008, 130, 6119–6130 CrossRef PubMed.
- J. P. S. Mowat, S. R. Miller, J. M. Griffin, V. R. Seymour, S. E. Ashbrook, S. P. Thompson, D. Fairen-Jimenez, A.-M. Banu, T. Düren and P. A. Wright, Inorg. Chem., 2011, 50, 10844–10858 CrossRef CAS PubMed.
- J. Gonzalez, R. N. Devi, P. A. Wright, D. P. Tunstall and P. A. Cox, J. Phys. Chem. B, 2005, 109, 21700–21709 CrossRef CAS PubMed.
- J. Gonzalez, R. N. Devi, D. P. Tunstall, P. A. Cox and P. A. Wright, Microporous Mesoporous Mater., 2005, 84, 97–104 CrossRef CAS.
- R. N. Devi, M. Edgar, J. Gonzalez, A. M. Z. Slawin, D. P. Tunstall, P. Grewal, P. A. Cox and P. A. Wright, J. Phys. Chem. B, 2004, 108, 535–543 CrossRef CAS.
- V. J. Carter, J. P. Kujanpää, F. G. Riddell, P. A. Wright, J. F. C. Turner, C. R. A. Catlow and K. S. Knight, Chem. Phys. Lett., 1999, 313, 505–513 CrossRef CAS.
- K. Sumida, C. M. Brown, Z. R. Herm, S. Chavan, S. Bordiga and J. R. Long, Chem. Commun., 2011, 47, 1157–1159 RSC.
- P. Canepa, N. Nijem, Y. J. Chabal and T. Thonhauser, Phys. Rev. Lett., 2013, 110, 026102(1) CrossRef PubMed.
- R. F. Code and N. F. Ramsey, Phys. Rev. A: At., Mol., Opt. Phys., 1971, 4, 1945–1959 CrossRef.
- B. Walaszek, A. Adamczyk, T. Pery, Y. P. Xu, T. Gutmann, N. D. Amadeu, S. Ulrich, H. Breitzke, H. M. Vieth, S. Sabo-Etienne, B. Chaudret, H. H. Limbach and G. Buntkowsky, J. Am. Chem. Soc., 2008, 130, 17502–17508 CrossRef CAS PubMed.
- V. I. Bakhmutov, Magn. Reson. Chem., 2004, 42, 66–70 CrossRef CAS PubMed.
- S. Macholl, J. Matthes, H. H. Limbach, S. Sabo-Etienne, B. Chaudret and G. Buntkowsky, Solid State Nucl. Magn. Reson., 2009, 36, 137–143 CrossRef CAS PubMed.
- F. Wehrmann, J. Albrecht, E. Gedat, G. J. Kubas, J. Eckert, H. H. Limbach and G. Buntkowsky, J. Phys. Chem. A, 2002, 106, 2855–2861 CrossRef CAS.
- F. Wehrmann, T. P. Fong, R. H. Morris, H.-H. Limbach and G. Buntkowsky, Phys. Chem. Chem. Phys., 1999, 1, 4033–4041 RSC.
- J. G. Vitillo, L. Regli, S. Chavan, G. Ricchiardi, G. Spoto, P. D. C. Dietzel, S. Bordiga and A. Zecchina, J. Am. Chem. Soc., 2008, 130, 8386–8396 CrossRef CAS PubMed.
- P. D. C. Dietzel, P. A. Georgiev, J. Eckert, R. Blom, T. Strassle and T. Unruh, Chem. Commun., 2010, 46, 4962–4964 RSC.
- M. T. Kapelewski, S. J. Geier, M. R. Hudson, D. Stück, J. A. Mason, J. N. Nelson, D. J. Xiao, Z. Hulvey, E. Gilmour, S. A. FitzGerald, M. Head-Gordon, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2014, 136, 12119–12129 CrossRef CAS PubMed.
- L. C. Lin, J. Kim, X. Q. Kong, E. Scott, T. M. McDonald, J. R. Long, J. A. Reimer and B. Smit, Angew. Chem., Int. Ed., 2013, 52, 4410–4413 CrossRef CAS PubMed.
- W. D. Wang, B. E. G. Lucier, V. V. Terskikh, W. Wang and Y. Huang, J. Phys. Chem. Lett., 2014, 5, 3360–3365 CrossRef CAS PubMed.
- B. E. G. Lucier, H. Chan, Y. Zhang and Y. Huang, Eur. J. Inorg. Chem., 2016, 2017–2024 CrossRef CAS.
- H. Lee, T. Polenova, R. H. Beer and A. E. McDermott, J. Am. Chem. Soc., 1999, 121, 6884–6894 CrossRef CAS.
- J. A. Rood, B. C. Noll and K. W. Henderson, Inorg. Chem., 2006, 45, 5521–5528 CrossRef CAS PubMed.
- T. Pham, K. A. Forrest, E. H. L. Falcao, J. Eckert and B. Space, Phys. Chem. Chem. Phys., 2016, 18, 1786–1796 RSC.
- Y. Lu, University of Western Ontario Electronic Thesis and Dissertation Repository, 2015, Paper 3237, http://ir.lib.uwo.ca/etd/3237.
Footnotes |
| † Electronic supplementary information (ESI) available: Full details of MOF synthesis, D2 loading, and SSNMR experiments. Illustrations of the MOFs, H2/D2 adsorption sites in MOF-74, additional 2H SSNMR spectra and simulations, tables, and powder XRD patterns are also included. See DOI: 10.1039/c6cc03205b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |