
Catalytic C–F bond activation of geminal difluorocyclopropanes by nickel(I) complexes via a radical mechanism†
Jan
Wenz
,
Christoph A.
Rettenmeier
,
Hubert
Wadepohl
and
Lutz H.
Gade
*
Anorganisch-Chemisches Institut, University of Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: lutz.gade@uni-heidelberg.de
First published on 13th November 2015
Abstract
Nickel(II) fluorido complexes bearing NNN-pincer ligands were found to be catalysts in the hydrodefluorination of geminal difluorocyclopropanes which undergo ring-opening to form the corresponding monofluoroalkenes in good yield and high Z-selectivities. Evidence for a radical based mechanism involving nickel(I) and nickel hydrido complexes as key intermediates was obtained in the corresponding stoichiometric reactions.
The activation and functionalization of C–F bonds is considered a major challenge in organometallic chemistry1 and has received growing attention due to the importance that organofluorine compounds have gained in recent years.2 The increasing demand for ways of introducing fluorine into new materials or into biologically active molecules has inspired the development of diverse synthetic strategies.1a
Hydrodefluorination (HDF) is regarded as a promising approach to access partially fluorinated building blocks from readily available fluorinated bulk chemicals.3 At present HDF is rarely used in preparative contexts but a range of transition metal catalysts have been described to date, including titanium,4 zirconium,5 rhodium,6 ruthenium,7 gold,8 palladium9 and nickel.1a,10
Despite the availability of several synthetic methods for the construction of geminal difluorocyclopropanes,11 their application as substrates in catalytic transformations has barely been examined. Very recently, Fu et al. reported a general and efficient Pd(0)/Pd(II)-catalyzed regioselective functionalization of geminal difluorocyclopropanes leading to 2-fluoroallylic amines, ethers, esters, and alkylation products with high Z-selectivities. Their contribution represents the first general application of geminal difluorocyclopropanes in this context.12
The pincer ligands used in this work13 are capable of stabilizing T-shaped nickel(I) complexes,14 which have been used to reduce prochiral geminal dichlorides and dibromides enantioselectively to the corresponding secondary halides in combination with a hydride source.14b A detailed mechanistic investigation of the reaction mechanism revealed a catalytic cycle that is based on the interplay between these nickel(I) complexes and corresponding nickel(II) hydrido species and involves the generation of α-halogen alkyl radicals.
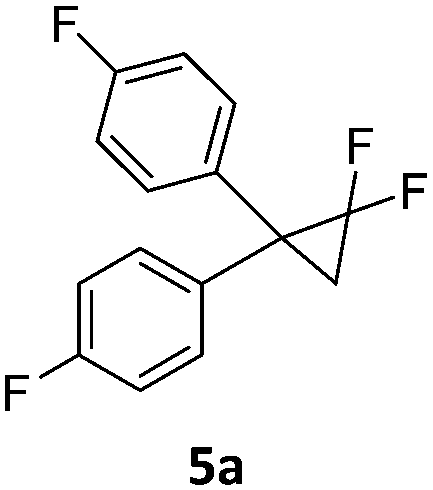
Here we report the activation of geminal difluorocyclopropanes by these nickel complexes inducing ring-opening of the cyclopropane which leads to fluoroalkenes with high Z-selectivity. The nickel(II) fluorido complexes 1a and 1b were used in the catalytic transformation. Salt metathesis of CsF or NH4F with the corresponding halogenido or hydroxo complexes in wet THF led to a clean formation of air and moisture stable fluorido complexes 1a,1b which were isolated and characterized (Scheme 1).15,16 The addition of water helps to dissolve the fluoride salts. Under these conditions the nickel fluorido complexes were found to be stable and no hydroxo species were formed. Notably, the 19F NMR resonances of the fluorido ligands are observed at unusually high field (−444.0 ppm (1a), −452.1 ppm(1b)) compared to the range reported in the literature (−160 ppm to −410 ppm).16,17
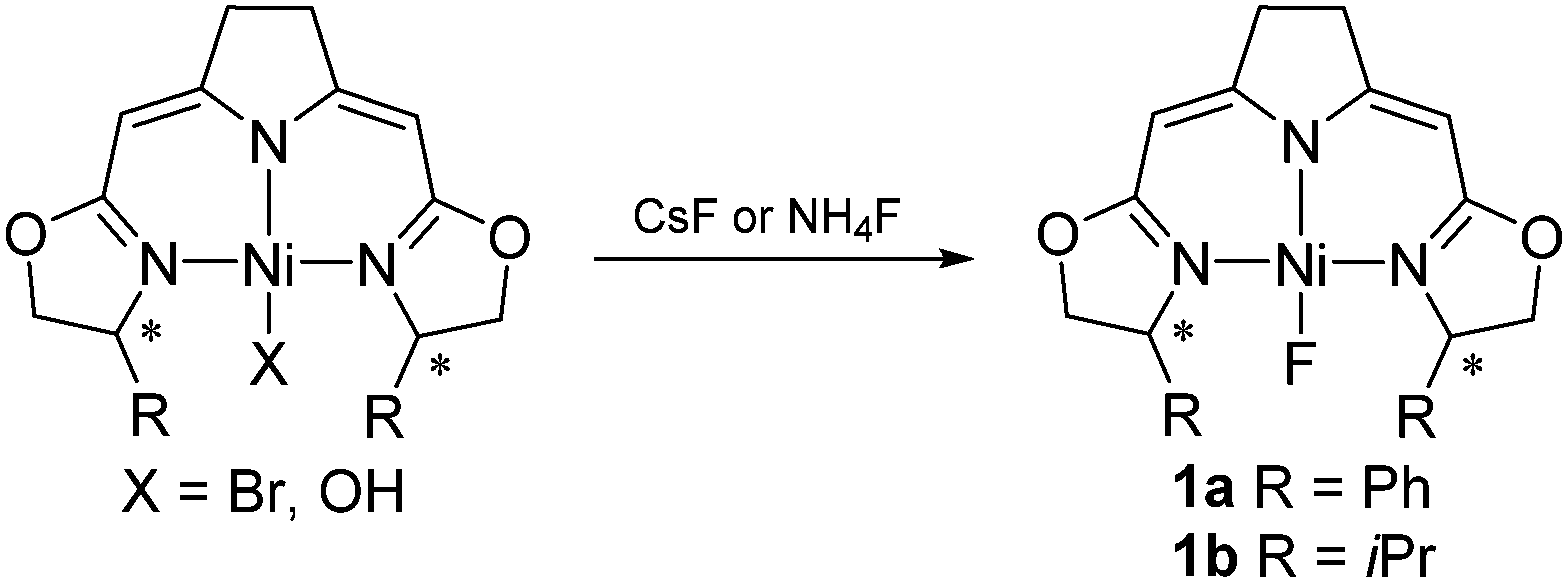 | ||
| Scheme 1 Synthesis of nickel fluorido complexes 1a and 1b. | ||
The high affinity of fluorine towards silicon3a and boron3c results in an increased reactivity of the fluorido complexes 1a and 1b with silanes and boranes in their conversion to the hydrido complexes 2a and 2b compared to the corresponding chlorido and bromido complexes.1a,18 The formation of the hydrido species (Scheme 2), which were previously shown to exist in a hydrogen pressure dependent equilibrium with the nickel(I) complexes (3a,3b), plays a crucial role in the catalytic hydrodefluorination. Generally, hydride transfer reagents such as PhSiH3 and Me2NHBH3 were found to be suitable stoichiometric hydride sources for the conversion of the fluorido to the hydrido complexes instead of the highly reactive LiEt3BH used in the previous catalytic hydrodehalogenations.14b
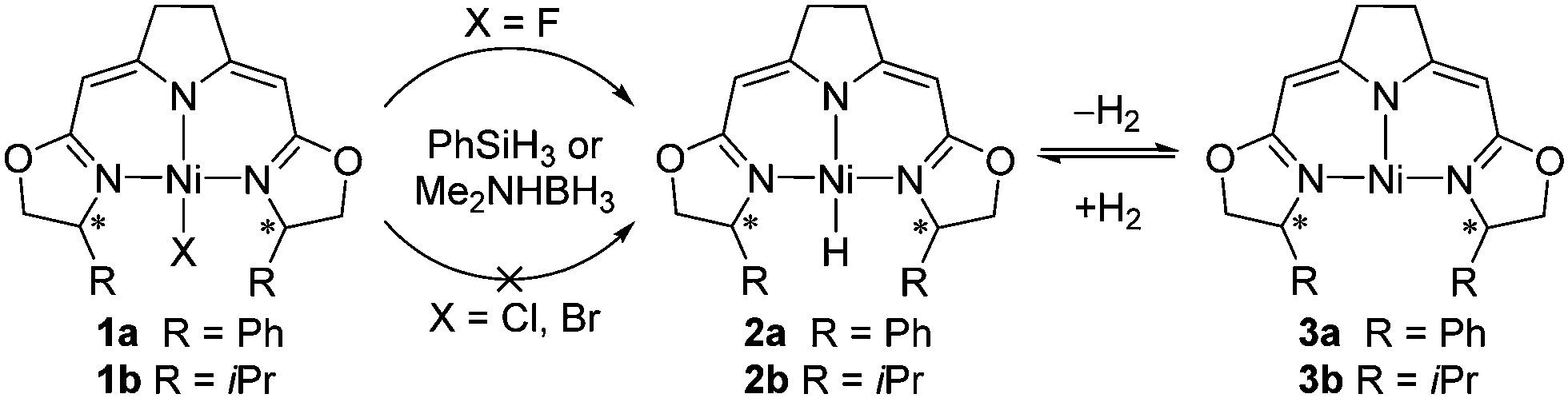 | ||
| Scheme 2 Conversion of the fluoridonickel complexes 1a and 1b with ammonia boranes and silanes to the corresponding hydrido complexes 2a and 2b and their H2-pressure dependent equilibrium with the T-shaped nickel(I) species. | ||
Single-crystal X-ray diffraction studies of 1b and 3b established small differences in the coordination of the pincer ligand to the nickel center for both oxidation states (Fig. 1).1 While 1b displays an almost ideal square planar coordination geometry, the absence of the fluorido ligand in 3b results in a slightly disordered T-shaped arrangement, which has already been shown for the corresponding complex 3a.14b The change in oxidation state is mainly reflected by an elongation of the central Ni–N(2) bond from 1.885(2) Å (1b) to 1.9301(18) Å (3b), which is attributed to the larger ionic radius of the nickel(I) center.
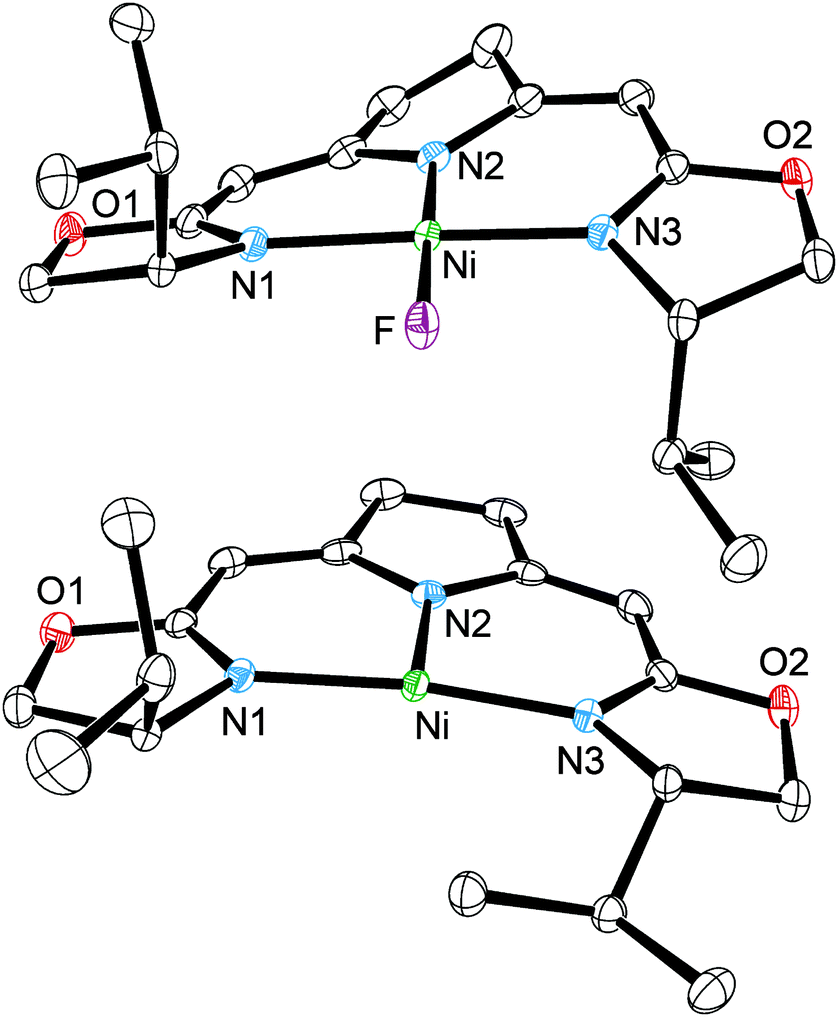 | ||
| Fig. 1 Molecular structures of 1b (top, only one of the two independent molecules is shown; cocrystallized toluene and hydrogen bonded H2O are not shown) and 3b (bottom). Hydrogen atoms are omitted for clarity. Selected bond length [Å] and angles [°]: (1b) Ni–F 1.8308(17), Ni–N(1) 1.8847(18), Ni–N(2) 1.885(2), Ni–N(3) 1.8884(18), F–Ni–N(1) 87.30(6), F–Ni–N(2) 178.96(6), F–Ni–N(3) 88.04(6), N(2)–Ni–N(1) 92.34(7), N(3)–Ni–N(1) 175.28(7); (3b) Ni–N(2) 1.9301(18), Ni–N(1) 1.8955(19), Ni–N(3) 1.8903(18), N(1)–Ni–N(2) 95.26(8), N(2)–Ni–N(3) 96.12(8), N(3)–Ni–N(1) 168.60(7). | ||
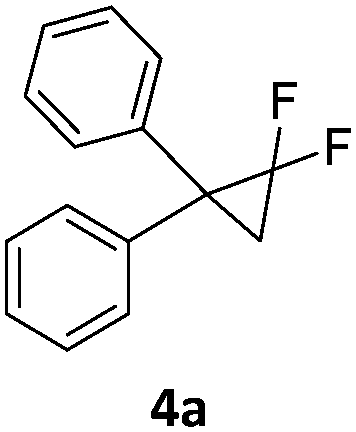
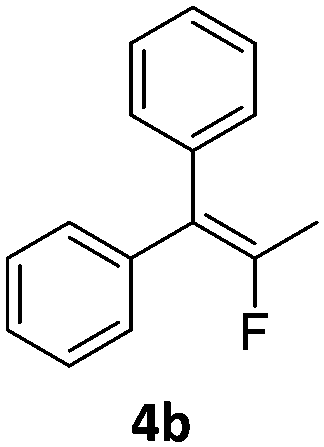
In the presence of a hydride source the nickel(II) fluorido complexes 1a,1b were able to catalytically activate 1,1-difluoro-2,2-diphenylcyclopropane above 60 °C leading to a C–C bond cleavage to form 1,1-diphenyl-2-fluoropropene (Scheme 3).
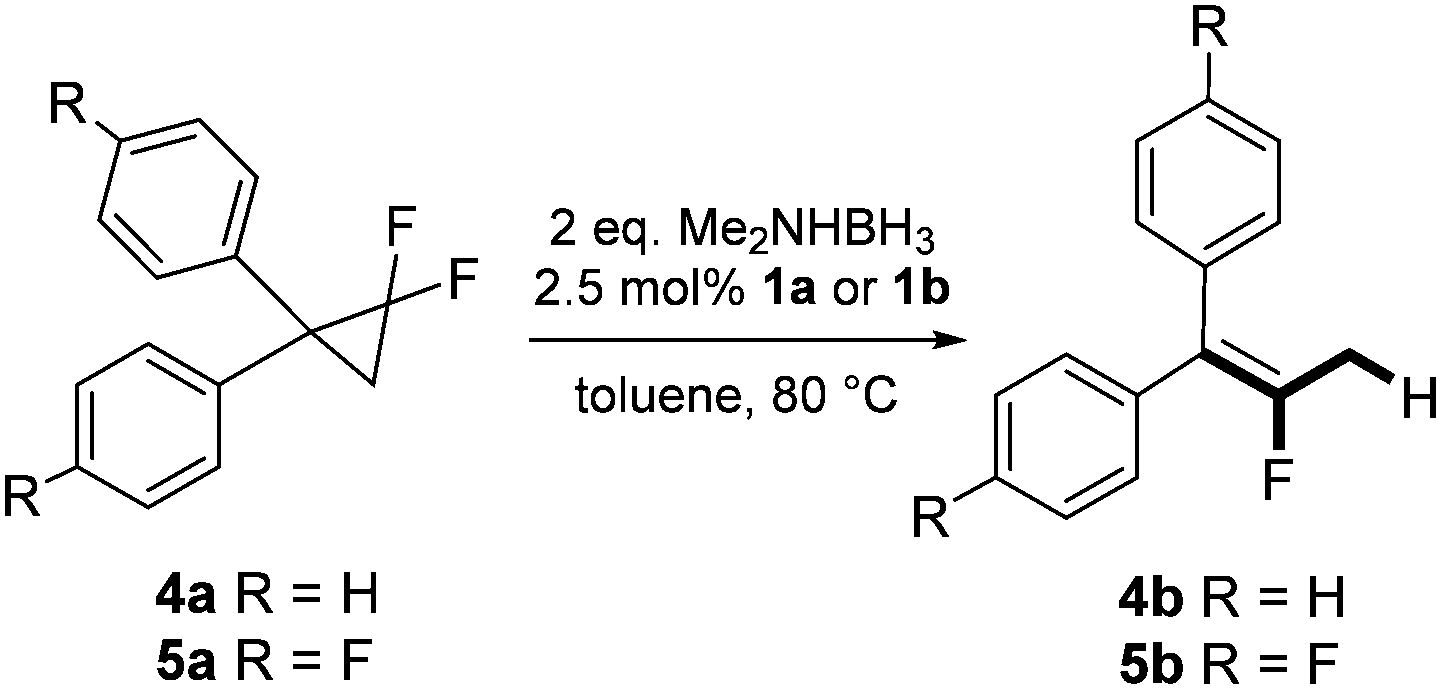 | ||
| Scheme 3 Catalytic hydrodefluorination of the 1,1-difluoro-2,2-diphenylcyclopropanes 4a and 4b. | ||
When the reaction was monitored by 19F NMR spectroscopy, the disappearance of the starting material [triplet resonance at −130.0 ppm (3JH,F = 8.7 Hz)] was accompanied by the appearance of a quartet at −97.4 ppm (3JH,F = 17.7 Hz) which correspond to the fluoroalkene 4b. The conditions of the reaction were optimized using the difluorocyclopropane derivative 5a. The best results were obtained with dimethylammonia borane as hydride source and the Ph-substituted nickel fluorido complex 1a in toluene as solvent at a temperature at 80 °C (ESI†). Under these optimized conditions a range of difluorocyclopropanes (4a–11a) was tested (Table 1). After a reaction time of 16 h, the complete conversion of the starting material had occurred in all cases and the formation of the corresponding fluoroalkenes with high Z-selectivity and yield was observed. Notably, the nickel system described in this work is capable of activating the sterically demanding 1,1′-disubstituted difluorocyclopropanes.
|
|
|||
|---|---|---|---|
| Substrate | Product | dra (Z/E) | Yield [%] (NMRa) |
| a Determined by 19F-NMR before work-up using 1,4-bis(trifluoromethyl)benzene as internal standard. | |||
|
|
— | 89 (99) (1a) |
| (96) (1b) | |||
|
|
— | 86 (99) (1a) |
| (88) (1b) | |||
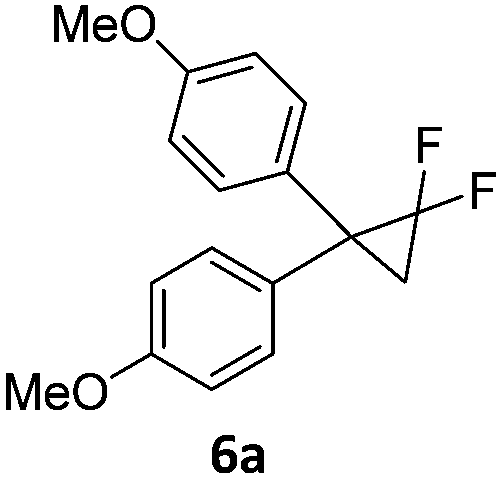
|
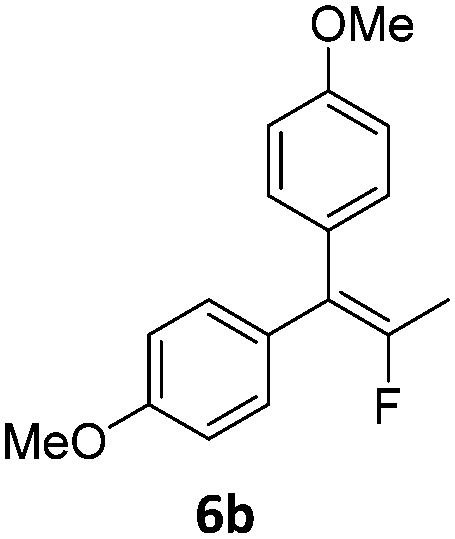
|
— | 92 (98) (1a) |
| (75) (1b) | |||
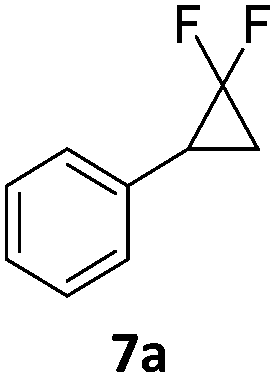
|
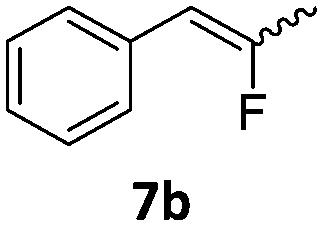
|
9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (1a) :1 (1a) |
67 (90) (1a) |
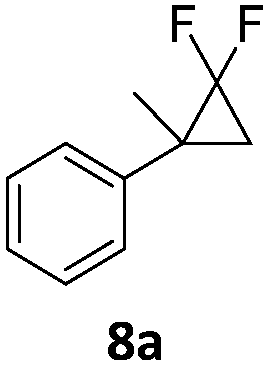
|
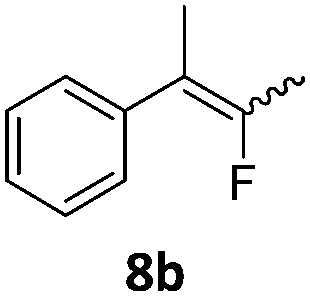
|
7:3 (1a) |
65 (74) (1a) |
| 6:4 (1b) |
|||
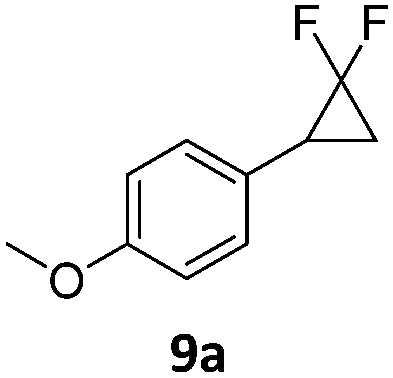
|
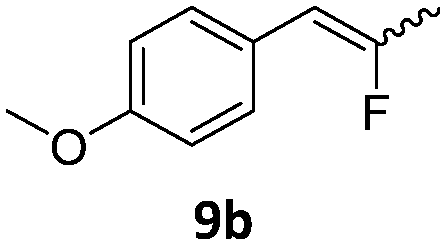
|
19:1 (1a) |
61 (78) (1a) |
| 13:1 (1b) |
(53) (1b) | ||
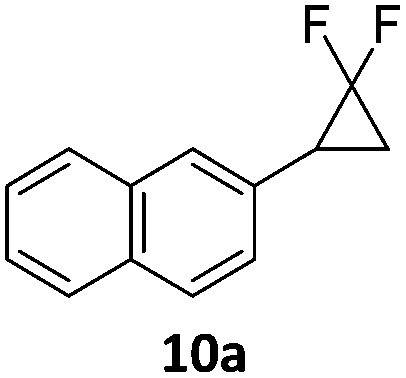
|
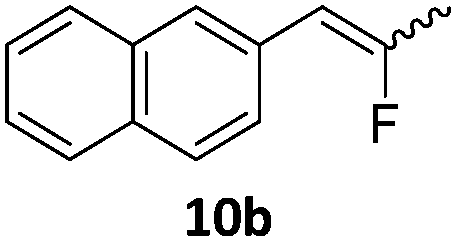
|
9:1 (1a) |
89 (96) (1a) |
| 9:1 (1b) |
(60) (1b) | ||
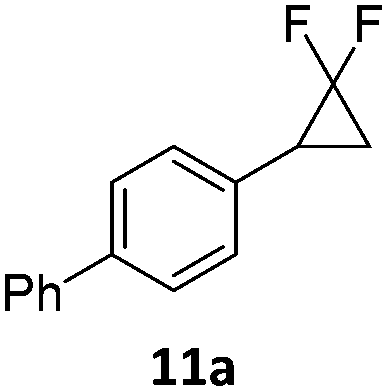
|
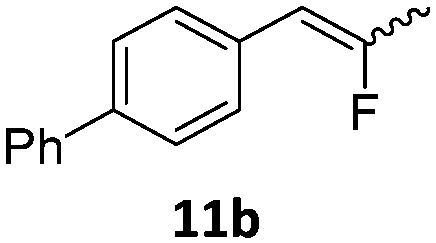
|
9:1 (1a) |
81 (99) (1a) |
| 9:1 (1b) |
(64) (1b) | ||
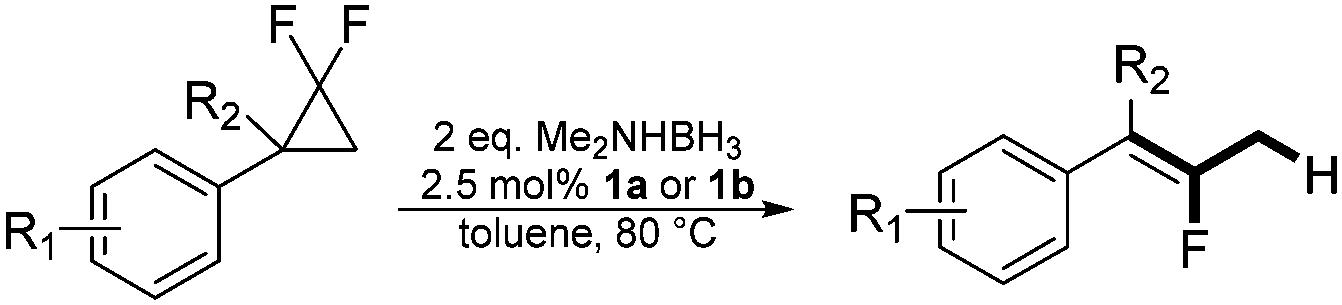
In the case of chiral geminal difluorocyclopropanes, which were employed as racemates, a mixture of both possible diastereomeric fluoroalkenes was obtained (entry 7a–11a) and the diastereomeric ratio was determined by 19F NMR spectroscopic analysis. The characteristic trans and cis hydrogen fluorine coupling constants of 3JH,F(trans) ≈ 45 Hz and 3JH,F(cis) ≈ 18 Hz were used to determine the configuration of the double bond of each isomer.19 In each case the Z-diastereomer was obtained as the major product with moderate to excellent diastereoselectivity. Since the observed product ratio was found to be independent of the conversion, the possibility of a kinetic resolution as the reason for preferred formation of one stereoisomer can be ruled out. DFT modelling of E- and Z-diastereomers revealed slightly greater stability of the latter, except for 8b (see ESI†).
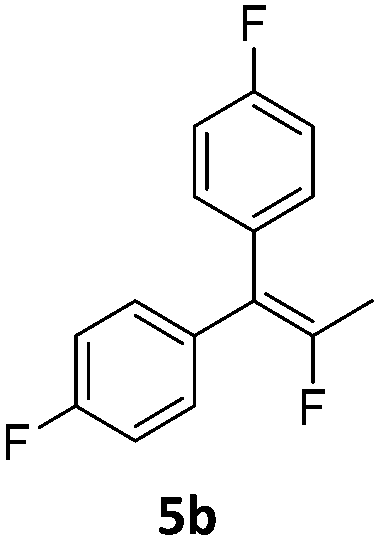
In analogy to the previously studied hydrodechlorinations, the activation of the C–F bonds by the nickel catalysts employed in this work is thought to involve the in situ generated nickel(I) species 3a,3b which abstract halogen atoms via one electron steps involving radical species. It was thus essential to probe whether the significantly stronger C–F bond could be activated by the isolated nickel(I) species. To this end, stoichiometric transformations involving the geminal difluorocyclopropane 5a were carried out. The reaction of 5a with one equiv. of the nickel(I) complex 3a at 80 °C in toluene led to a 1:1 mixture of the starting material and diphenylallene 12 along with one equivalent of the fluorido complex 1a. On the other hand, reaction with two molar equivalents of 3a gave a near quantitative formation of the reaction product 12 (Scheme 4).‡ The exclusive observation of the doubly defluorinated product indicated that the second defluorination step occurs more rapidly than the initial fluorine atom abstraction.
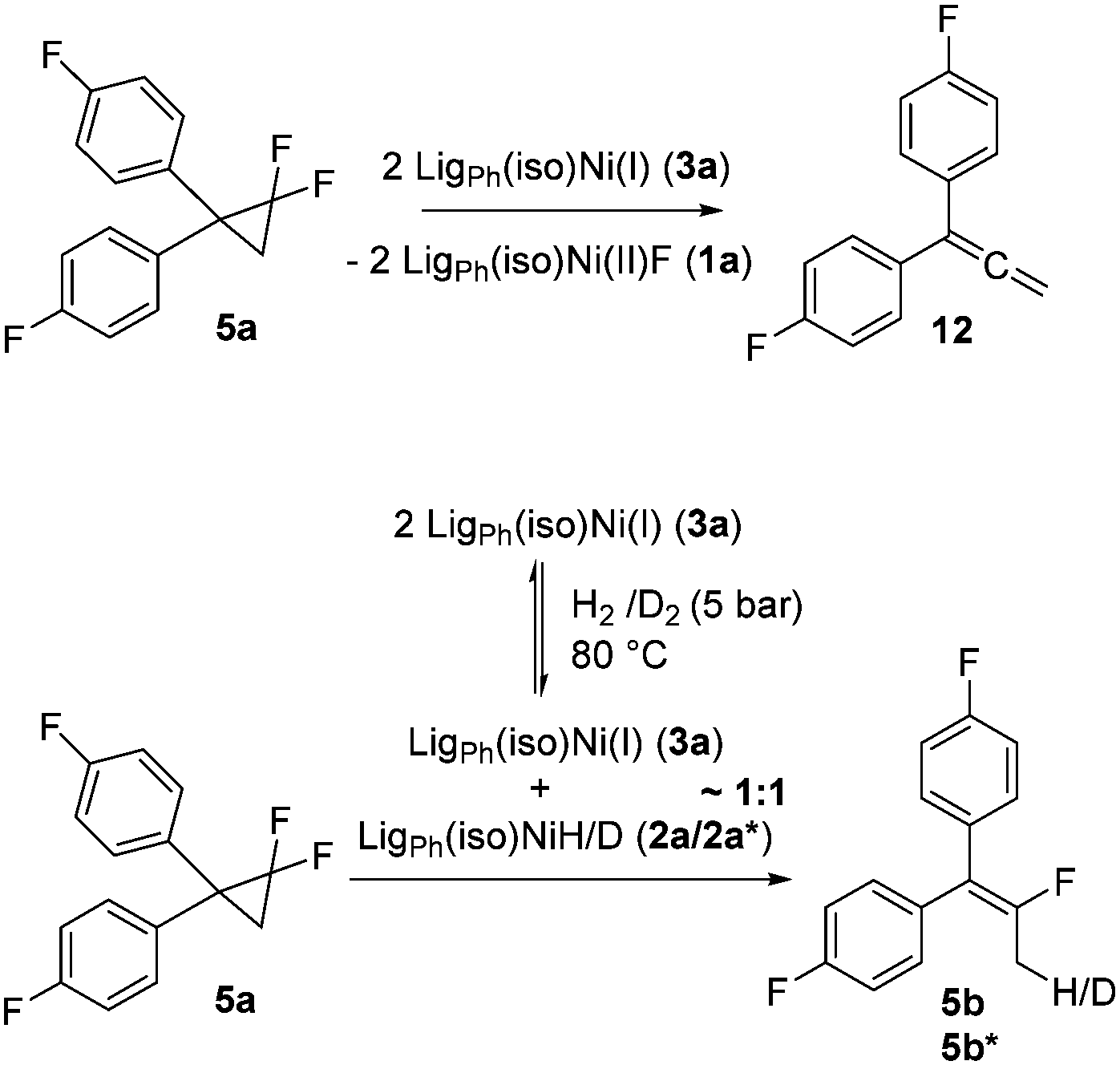 | ||
| Scheme 4 Stoichiometric reactions of 5a with the nickel complexes 2a and 3a. Top: Formation of a 1,1′-diphenylallene derivative via double defluorination of 5a. Bottom: Monodefluorination and subsequent hydrogen/deuterium atom transfer in the presence of an in situ generated mixture of 2a/2a* and 3a. | ||
In order to probe the reactivity of the elusive monodefluorinated intermediate and to obtain insight into the reaction paths leading to the fluoroalkene products of the catalytic process discussed above, the solution of the Ni(I) complex 3a was placed under 5 bar of hydrogen at 80 °C to generate in situ a ratio of roughly 1:1 between both nickel hydride species 2a and the nickel(I) complex 3a (which are in equilibrium with each other under these conditions). Performing the same stoichiometric transformation under these conditions gave the hydrodefluorination product 5b exclusively. The H atom transfer from the hydrido complex 2a onto an allylic (radical) intermediate is thus kinetically favoured over a second F atom abstraction by the nickel(I) species 3a. If, instead of hydrogen, deuterium was used the exclusive formation of the mono deuterated product 5b* was observed. Due to the reaction temperature of 80 °C under which these transformations occur, an investigation of the nature of the intermediate species by the usual radical traps was not possible.
However, the observations are generally consistent with a mechanism which is similar to the one established previously for the hydrodehalogenation of geminal dichlorides: in a first step the nickel(I) complex activates the geminal difluorocyclopropane 4a–11a by homolytic C–F cleavage and subsequent ring-opening20 liberating the monofluoroallylic radical species 13 which may be either metal-stabilized or dissociated (Scheme 5). The latter is transformed to the corresponding fluoroalkene 4b–11bvia hydrogen atom abstraction from the nickel hydrido complex 2 (as demonstrated in the stoichiometric reaction discussed above) regenerating the nickel(I) species 3. The rapid conversion of the fluorido complex 1, formed in the first defluorination step, to the hydrido complex 2 by the hydride source closes the catalytic cycle.
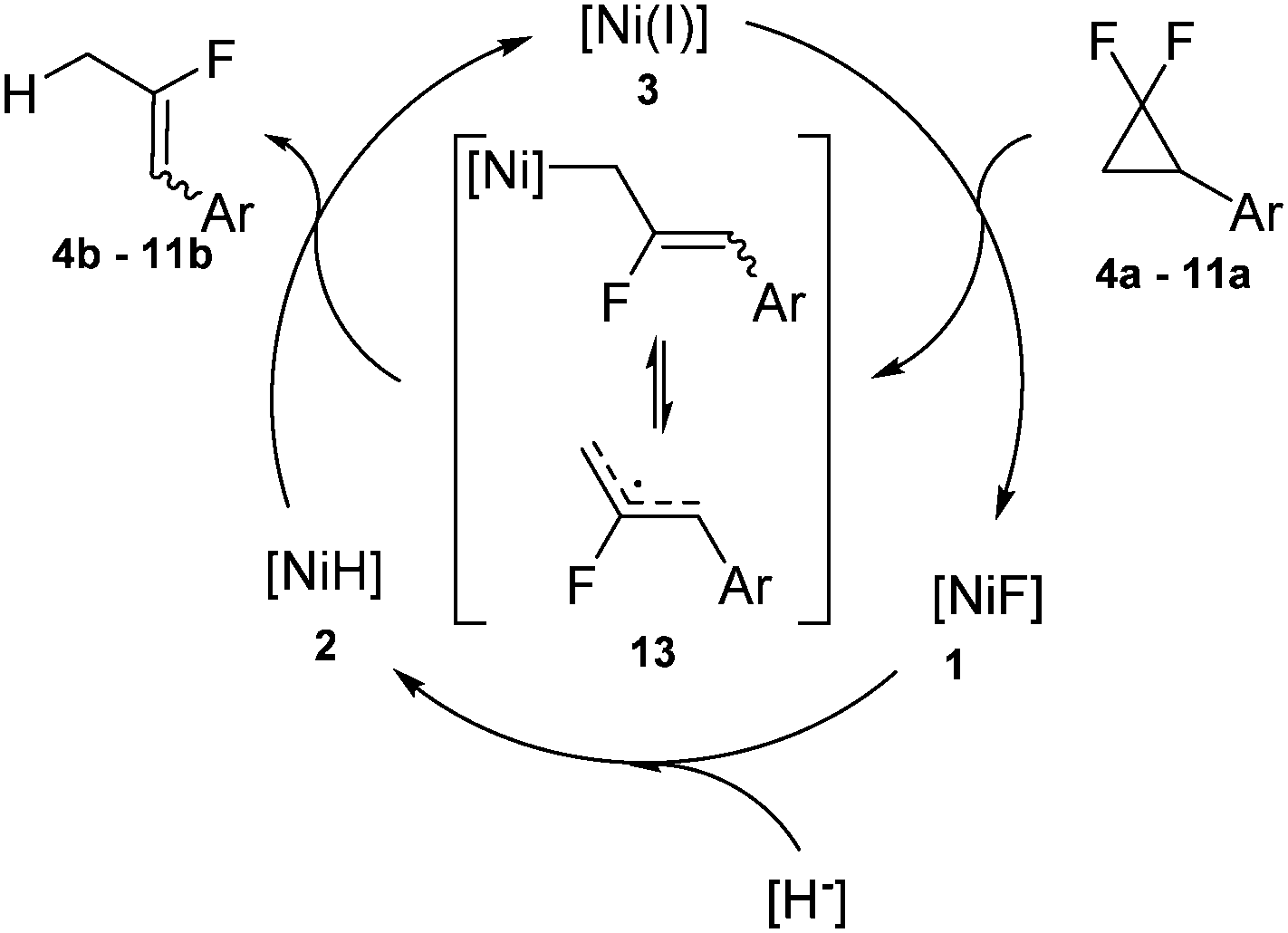 | ||
| Scheme 5 Proposed mechanism for the catalytic cycle of the hydrodehalogenation of geminal difluorides. | ||
To further support the mechanistic proposal in which a C–F bond is activated by the nickel(I) species in the initial step of the catalytic cycle, we probed to see whether non-constrained geminal fluorides would be disposed to defluorination under these conditions. Indeed, the reaction of perfluorinated decalin 14 with 10 equiv. of the isolated nickel(I) species at room temperature led to the quantitative formation of perfluorinated naphthalene 15 as well as the nickel fluorido complex 1a (Scheme 6). The formation of the unsaturated compound during this reaction reflects the general reactivity pattern observed for the dehalogenation of dihalogenated alkanes by Ni(I) (TMC) complexes21 and, therefore, supports the proposed occurrence of radical intermediates in the defluorination reaction of difluorocyclopropanes presented in this work.
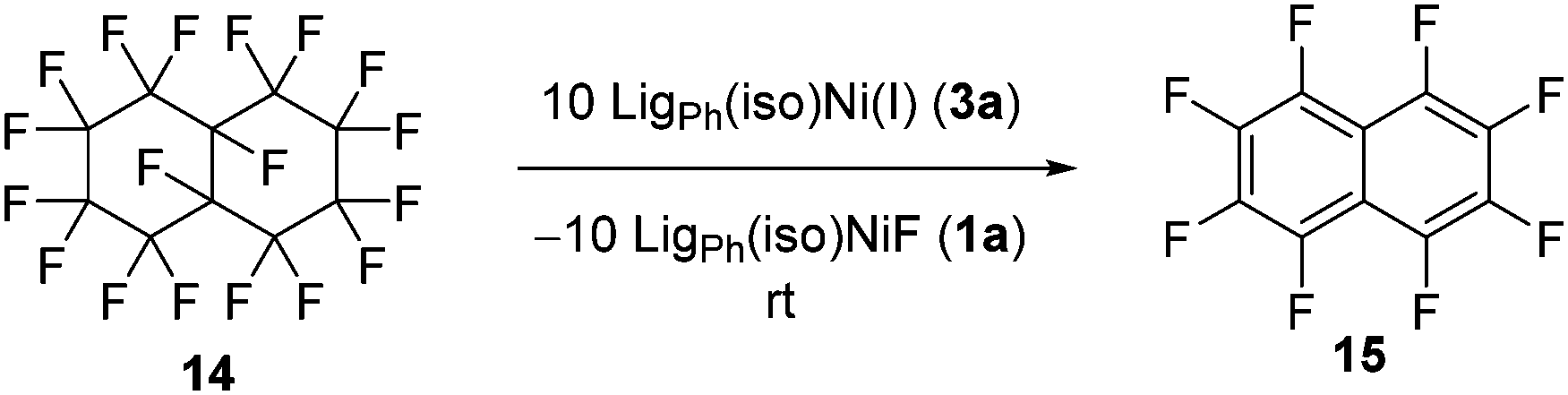 | ||
| Scheme 6 Stoichiometric defluorination of 14 by 3a. | ||
The results of this work demonstrate that the previously developed nickel(I/II) system, which was found to be catalytically active for the hydrodechlorination and -bromination of corresponding geminal dihalides is also capable of activating the geminal difluorocyclopropanes. The products, most probably formed via an analogous radial mechanism are vinylic fluorides which are obtained in high yield and Z-selectivity.
We thank Alexander Ahrens and Steffen Ott for experimental support. We acknowledge funding by the Deutsche Forschungsgemeinschaft (Ga 488/9-1) as well as the University of Heidelberg.
Notes and references
- (a) M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328 CrossRef PubMed; (b) T. Braun, Organometallics, 2012, 31, 1213 CrossRef CAS; (c) J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373 CrossRef CAS; (d) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed.
- (a) V. Gouverneur and K. Seppelt, Chem. Rev., 2015, 115, 563 CrossRef CAS PubMed; (b) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef CAS PubMed; (c) P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) M. K. Whittlesey and E. Peris, ACS Catal., 2014, 4, 3152 CrossRef CAS; (b) E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333 CrossRef CAS PubMed; (c) C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946 CrossRef CAS PubMed; (d) G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 48, 1546 CrossRef CAS PubMed; (e) J. Xiao, J. Wu, W. Zhao and S. Cao, J. Fluorine Chem., 2013, 146, 76 CrossRef CAS.
- M. F. Kuehnel, P. Holstein, M. Kliche, J. Krüger, S. Matthies, D. Nitsch, J. Schutt, M. Sparenberg and D. Lentz, Chem. – Eur. J., 2012, 18, 10701 CrossRef CAS PubMed.
- B. L. Edelbach, A. K. Fazlur Rahman, R. J. Lachicotte and W. D. Jones, Organometallics, 1999, 18, 3170 CrossRef CAS.
- (a) T. Braun and F. Wehmeier, Eur. J. Inorg. Chem., 2011, 613 CrossRef CAS; (b) O. Ekkert, S. D. A. Strudley, A. Rozenfeld, A. J. P. White and M. R. Crimmin, Organometallics, 2014, 33, 7027 CrossRef CAS.
- S. P. Reade, M. F. Mahon and M. K. Whittlesey, J. Am. Chem. Soc., 2009, 131, 1847 CrossRef CAS PubMed.
- J.-H. Zhan, H. Lv, Y. Yu and J.-L. Zhang, Adv. Synth. Catal., 2012, 354, 1529 CrossRef CAS.
- D. Breyer, T. Braun and A. Penner, Dalton Trans., 2010, 39, 7513 RSC.
- Selected references for hydrodefluorination reactions at nickel: (a) W. Zhao, J. Wu and S. Cao, Adv. Synth. Catal., 2012, 354, 574 CrossRef CAS; (b) J. Wu and S. Cao, ChemCatChem, 2011, 3, 1582 CrossRef CAS; (c) P. Fischer, K. Götz, A. Eichhorn and U. Radius, Organometallics, 2012, 31, 1374 CrossRef CAS; (d) N. Y. Adonin and V. F. Starichenko, J. Fluorine Chem., 2000, 101, 65 CrossRef CAS; (e) A. Arévalo, A. Tlahuext-Aca, M. Flores-Alamo and J. J. García, J. Am. Chem. Soc., 2014, 136, 4634 CrossRef PubMed.
- (a) L. Li, F. Wang, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2013, 52, 12390 CrossRef CAS PubMed; (b) F. Wang, T. Luo, J. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K. S. Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153 CrossRef CAS PubMed; (c) M. Fedoryński, Chem. Rev., 2003, 103, 1099 CrossRef PubMed; (d) W. R. Dolbier and M. A. Battiste, Chem. Rev., 2003, 103, 1071 CrossRef CAS PubMed; (e) R. Eujen and B. Hoge, J. Organomet. Chem., 1995, 503, C51 CrossRef CAS; (f) G. A. Wheaton and D. J. Burton, J. Fluorine Chem., 1977, 9, 25 CrossRef CAS.
- J. Xu, E.-A. Ahmed, B. Xiao, Q.-Q. Lu, Y.-L. Wang, C.-G. Yu and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 8231 CrossRef CAS PubMed.
- (a) F. Konrad, J. Lloret Fillol, C. Rettenmeier, H. Wadepohl and L. H. Gade, Eur. J. Inorg. Chem., 2009, 4950 CrossRef CAS; (b) C. Mazet and L. H. Gade, Chem. – Eur. J., 2003, 9, 1759 CrossRef CAS PubMed.
- (a) C. A. Rettenmeier, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2015, 54, 4880 CrossRef CAS PubMed; (b) C. Rettenmeier, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2014, 20, 9657 CrossRef CAS PubMed.
- N. M. Doherty and N. W. Hoffmann, Chem. Rev., 1991, 91, 553 CrossRef CAS.
- (a) J. Cámpora, I. Matas, P. Palma, C. Graiff and A. Tiripicchio, Organometallics, 2005, 24, 2827 CrossRef; (b) B. C. Fullmer, H. Fan, M. Pink, J. C. Huffman, N. P. Tsvetkov and K. G. Caulton, J. Am. Chem. Soc., 2011, 133, 2571 CrossRef CAS PubMed; (c) L. M. Martínez-Prieto, C. Melero, D. del Río, P. Palma, J. Cámpora and E. Álvarez, Organometallics, 2012, 31, 1425 CrossRef.
- J. A. Hatnean, M. Shoshani and S. A. Johnson, Inorg. Chim. Acta, 2014, 422, 86 CrossRef CAS.
- T. Schaub, M. Backes and U. Radius, Eur. J. Inorg. Chem., 2008, 2680 CrossRef CAS.
- W. R. Dolbier, Guide to Fluorine NMR for Organic Chemists, John Wiley & Sons, Inc., 2008 Search PubMed.
- T. Nihei, T. Hoshino and T. Konno, Org. Biomol. Chem., 2015, 13, 3721 CAS.
- (a) M. S. Ram, A. Bakac and J. H. Espenson, Inorg. Chem., 1988, 27, 4231 CrossRef CAS; (b) J. H. Espenson, M. S. Ram and A. Bakac, J. Am. Chem. Soc., 1987, 109, 6892 CrossRef CAS; (c) M. S. Ram, A. Bakac and J. H. Espenson, Inorg. Chem., 1988, 27, 2011 CrossRef CAS.
- K. Maruyama and H. Imahori, J. Org. Chem., 1989, 54, 2692 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, NMR, crystal data and structure refinement. CCDC 1427461–1427465. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc08950f |
| ‡ The product 1,1-diphenylallene was synthesized separately according to literature procedure (Doering–LaFlamme allene synthesis) to verify its formation during the reaction.22 |
| This journal is © The Royal Society of Chemistry 2016 |