
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gelation by supramolecular dimerization of mono(urea)s†
Abigail E.
Hooper
,
Stuart R.
Kennedy
 ,
Christopher D.
Jones
and
Jonathan W.
Steed
*
,
Christopher D.
Jones
and
Jonathan W.
Steed
*
Department of Chemistry, University of Durham, South Road, DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk; Fax: +44 (0)191 384 4737; Tel: +44 (0)191 334 2085
First published on 30th October 2015
Abstract
Mono-ureido salicylic acid derivatives form hydrogen bonded dimers that mimic the gelation behaviour of covalently linked bis(urea)s. The 5-ureido salicylic acid isomers are much more effective gelators because of the lowered conjugation and less planar geometry, resulting in stronger urea⋯urea α-tape hydrogen bonding interactions.
Gel phase soft materials based on supramolecular interactions between low molecular weight gelators (LMWG) have become extremely popular in the past decade. These versatile media have applications1,2 in organocatalysis,3,4 wound healing,5 drug delivery,6,7 as templates and supports for nanomaterials,8–10 and in pharmaceutical polymorph screening and control.11–15 LMWG represent a very broad class of compounds which have in common the ability to form high aspect ratio fibres by rapid unidirectional grown of a solid, sample-spanning network. Gels are formed in a wide range of solvents including water (hydrogels) and both polar and non-polar organic liquids (organogels). The reversibility of the supramolecular interactions between the small molecule gelators means that gels derived from LMWG are usually thermally reversible and their materials properties, particularly their rheological characteristics, can be manipulated by light, changes in pH or by chemical triggers such as metal cations and hydrogen bond acceptor anions.16–21 Common LMWG are based on chemical functionalities capable of forming one dimensional assemblies including nucleobases, amides and ureas.21–23 Most LMWG incorporate multiple functional groups that associate in a directional way in order to increase the strength of the fibres by multivalent supramolecular interactions. In urea-based systems most gelators (with some exceptions24–27) contain at least two urea groups.25,28–35 We now report a novel approach to effective urea-based gelators in which two urea functional groups are linked by a supramolecular hydrogen bonded motif instead of by a covalent bond. The interactions involved in the assembly of the mono(urea) LMWG into a dimer and the subsequent assembly of the hydrogen bonded dimers into a fibre are orthogonal to one another, and the different supramolecular homosynthons form with high fidelity, allowing effective gelation. The approach is related to the hexagonal isophthalic acid derived assemblies reported by Potluri and Hamilton.24
Gelator synthesis. A series of mono(urea)s based on 4- and 5-aminosalycylic acid were synthesised from reaction of the respective aminosalycylic acid with the appropriate isocyanate to give compounds 1a–d and 2a–d in good yield. A search of the CSD shows that the salicylic acid functionality is almost invariably planar because of strong intramolecular OH⋯O hydrogen bonding and tends to form hydrogen bonded carboxylic acid dimers via an R22(8) hydrogen bonded ring motif,36 as in the structure of 5-aminosalycylic acid itself and 17 out of 24 related derivatives.37 Based on the CSD evidence the supramolecular synthons38 formed by the salicylic acid and urea functional groups might be expected to be orthogonal to one another and hence self-sorting in their assembly behaviour.
Gelation. Gelation tests on compounds of type 1 and 2 were carried out at 1 mg mL−1 in a wide range of solvents. Samples were sonicated for 30 seconds, then heated gently to dissolve the solid, and subsequently allowed to cool to room temperature. Most gels formed within a few minutes while for some samples crystallisation occurred over an approximately 24 hour period. Full results are tabulated in the Table S1 (ESI†). The 5-ASA derivatives of type 2 proved to be surprisingly efficient gelators despite the presence of only a single urea group. The unbranched chain members of the series 2a, c and d form gels to some degree in 1,2,4-trichlorobenzene, 1,2-dibromoethane, 1,2-dichlorobenzene, 1,3-dichlorobenzene and nitrobenzene (see Fig. S1, ESI†). The gels form within a few minutes of cooling and are all opaque suggesting a relatively large particle size. Gels formed in 1,2,4-trichlorobenzene collapsed after 30 min, while 1,2-dibromoethane and 1,3-dichlorobenzene gave somewhat stronger gels, although these also collapsed after a few hours. The strongest and most optically transparent gels formed in 1,2-dichlorobenzene and nitrobenzene, which proved to be stable at least over a period of weeks. In comparison with the unbranched gelators, the isopropyl derivative 2b formed only partial gels in just two solvents and these materials collapsed immediately. It is possible that the increased steric bulk of the isopropyl group hinders urea self-assembly and hence disfavours gel formation.
The 4-ASA derivatives of type 1 proved to be more soluble than their 5 ASA counterparts and did not exhibit significant gelation behaviour. In only one case was a gel-like material obtained. Slow cooling of 1a in nitromethane gave an opaque, white self-supporting solid with a critical gelation concentration of 1% w/v. SEM analysis of the resulting xerogel showed it to comprise a mixture of fibrous and crystalline material (see Fig. S1 and S2, ESI†).
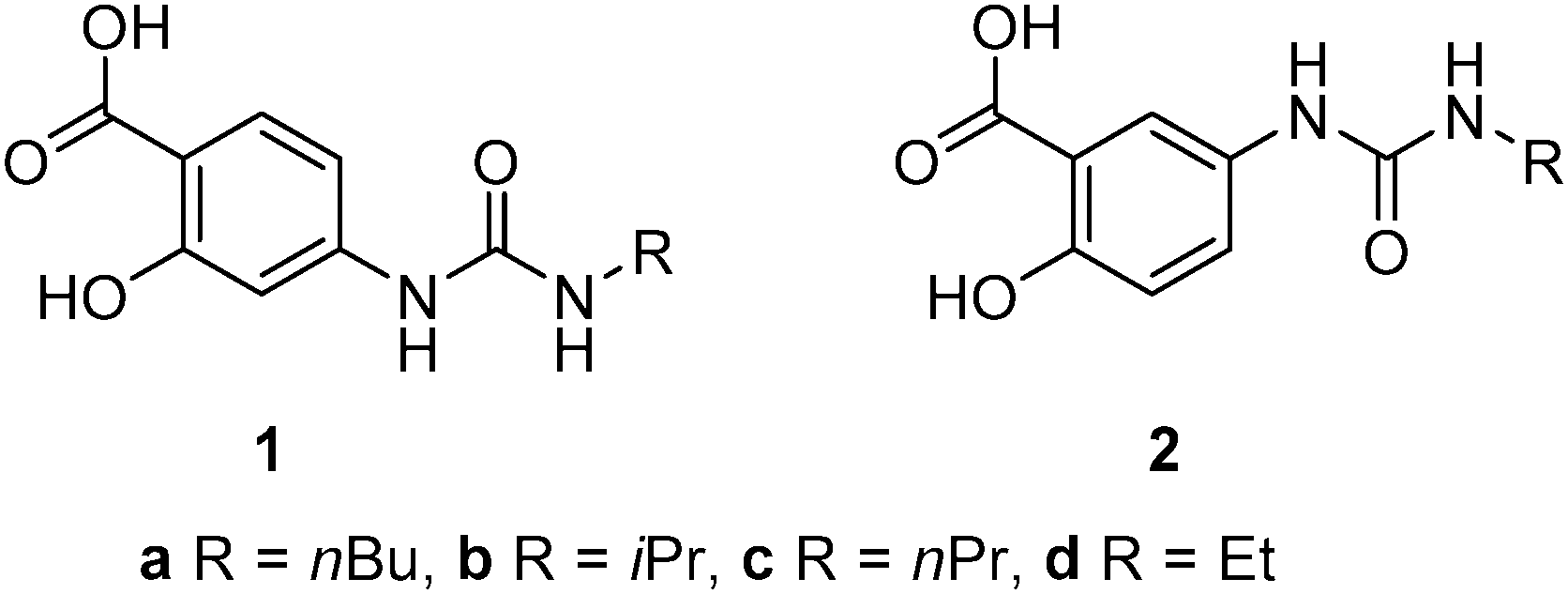
Representative gels were dried in a vacuum desiccator for 24 hours to give xerogels and then coated with either chromium or platinum and imaged by SEM. The microscope images showed that the xerogels adopt the expected fibrous morphology. Fibre widths and bunching vary from sample to sample but are consistent with other semi-transparent gels.39 A representative image of the xerogel of n-butyl compound 2a obtained from nitrobenzene is shown in Fig. 1a and contrasts with the xerogel of ethyl analogue 2d, which exhibits a mixture of shorter, blockier and more well defined crystalline tapes and long, thin and more curved fibres (Fig. 1b). The shorter ethyl substituent may result in more facile recrystallization of some of the sample during the drying process. SEM images of nitromethane xerogels of 1a, the only 4-ASA derived gel, show a significant difference from the 5-ASA versions, with wide crystalline aggregates of fibres forming sheets 1 μm across (Fig. S4, ESI†). All the fibres are distinctly larger than those of 1a and 1d in the same solvent. This may account for the more crystalline, opaque appearance of the gel. Gels of 2a, 2c and 2d were characterised by oscillatory stress sweep rheometry to probe their viscoelastic properties in several solvents. The solid-like character of the materials was manifest in the fact that the G′ (storage modulus) remains well over an order of magnitude higher than the viscous modulus G′′ up to the yield stress of the gel. The relatively weak nature of the gels of compound 2a in 1,2-dichlorobenzene is apparent in the fairly low value of G′ of ca. 1000 Pa at low stress, and the low yield stress of around 20 Pa, for example (Fig. 2a). This gel exhibits weak strain overshoot characterised by a rise on G′′ with increasing stress. This viscoelastic behaviour is indicative of close-packed weakly bonded particles forming a structure that resists deformation just prior to breaking of interactions and flow occurring.40,41 Elastic moduli were found to be highly solvent dependent and to increase with increasing chain length. Hence, 1,2-dibromoethane gels of the butyl compound 2a proved quite robust with G′ reaching ca. 10 kPa (Fig. 2b, see Fig. S5, ESI†).
 | ||
| Fig. 1 SEM images of (a) xerogel of 2a obtained from nitrobenzene and (b) xerogel of 2d obtained from nitromethane. | ||
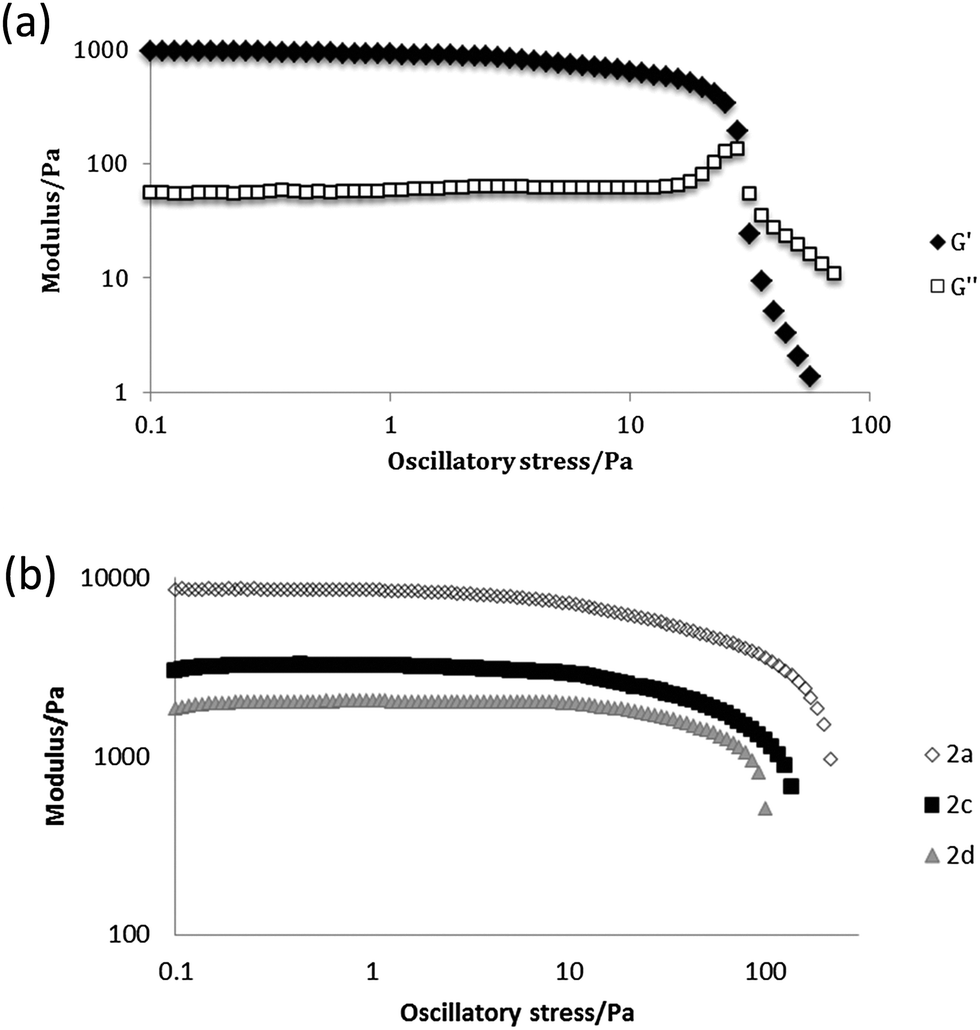 | ||
| Fig. 2 (a) Stress sweep rheometry for a 1% w/v gel of 2a in 1,2-dichlorobenzene. (b) comparison of the evolution of G′ in 1,2-dibromoethane as a function of chain length. | ||
Single crystals suitable for X-ray crystallography were obtained for compounds 1a, 1c and 1d by recrystallization from ethyl acetate, acetone and 2-butanone, respectively. The sample of 1d proved to be of very low quality and is not reported herein but a qualitative structure solution confirmed the packing is similar to the other two compounds of type 1. The structures of 1a and 1c are of high precision. The structures of the 4-ASA derivatives of type 1 all exhibit the common head-to-head carboxylic acid dimer motif,42 with approximate co-planarity of the carboxylic acid group and aryl ring enforced by an intramolecular OH⋯O interaction from the phenolic OH group to the acid carbonyl functionality, Fig. 3. The high acidity of the salicylic acid moiety results in short O⋯O distances across the R22(8) hydrogen bonded ring of 2.61–2.65 Å. The urea moieties are not precisely co-planar with the salicylic acid ring although there is a relatively long intramolecular ArCH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bond, as is the case with electron withdrawing diarylureas.43,44 In 1a the urea groups form a twisted R12(6) urea α-tape hydrogen bonded chain that is independent of the acid dimer hydrogen bonding. This urea motif, particularly in an antiparallel arrangement in covalently linked bis(urea)s, has been linked to the ubiquitous gel-forming ability of these compounds.28,39,45 However, in 1a, each hydrogen bonded dimer links into two different twisted α-tapes to give an overall 2D hydrogen bonded network based on two crystallographically independent molecules. In 1c, in contrast, the two urea α-tapes formed by each hydrogen bonded dimer are approximately coplanar and align antiparallel to give a 1D fibre, mimicking the arrangement found in covalently bonded bis(ureas).
C hydrogen bond, as is the case with electron withdrawing diarylureas.43,44 In 1a the urea groups form a twisted R12(6) urea α-tape hydrogen bonded chain that is independent of the acid dimer hydrogen bonding. This urea motif, particularly in an antiparallel arrangement in covalently linked bis(urea)s, has been linked to the ubiquitous gel-forming ability of these compounds.28,39,45 However, in 1a, each hydrogen bonded dimer links into two different twisted α-tapes to give an overall 2D hydrogen bonded network based on two crystallographically independent molecules. In 1c, in contrast, the two urea α-tapes formed by each hydrogen bonded dimer are approximately coplanar and align antiparallel to give a 1D fibre, mimicking the arrangement found in covalently bonded bis(ureas).
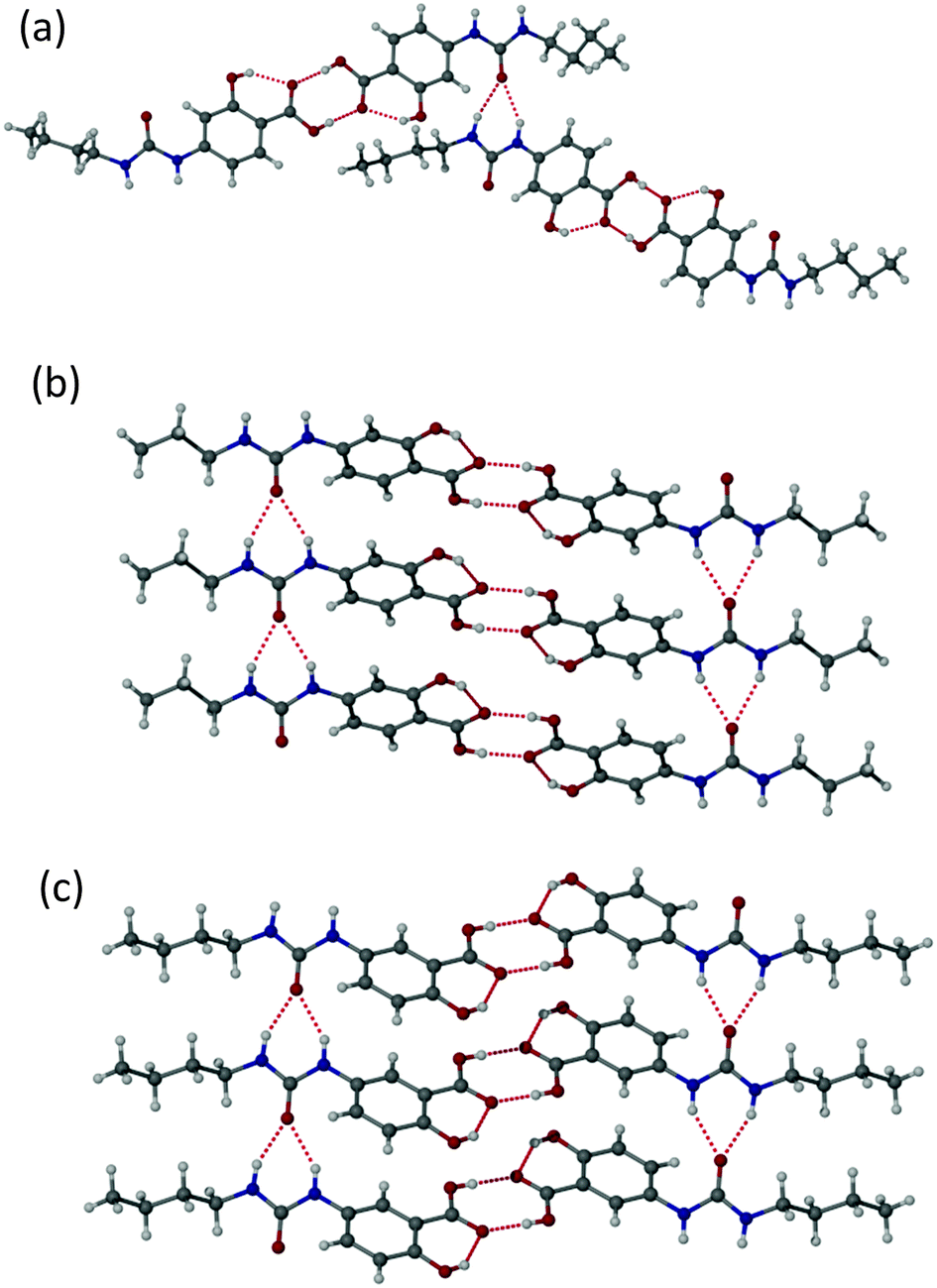 | ||
| Fig. 3 (a) Twisted 2D urea hydrogen bonding in 1a; (b) parallel packing in 1c with relatively long NH⋯O interactions; (c) short NH⋯O interactions in the gelator 2a. Selected hydrogen bonding distances (Å) for compound 1a: acid dimer O⋯O 2.606(2), 2.634(2); urea tape N⋯O 2.986(3), 2.880(2), 2.814(2) and 3.072(2), compound 1c, O⋯O 2.892(3), N⋯O 2.911(3), 2.892(3); compound 2a, O⋯O 2.629(6); N⋯O 2.797(7), 2.816(7). | ||
The 5-ASA derivative 2a was also characterised by single crystal X-ray crystallography using a sample obtained by recrystallization from methanol, Fig. 3c. As with the compounds of type 1, this compound also exhibits a head-to-head carboxylic acid dimer arrangement and, as with 1c, each dimer stacks with its neighbours to give an antiparallel pair of urea α-tape interactions running throughout the crystal. Notably the urea NH⋯O distances of 2.80–2.82 Å are at the lower end of the typical range found for this interaction44 and are considerably shorter than the in 1a and 1c (the interactions in 1a are unsymmetrical at 2.81/3.07 and 2.88/2.98 Å, while those in 1c are symmetrical at 2.89–2.91 Å). This short urea⋯urea distance is correlated with the much more significant out-of-plane orientation of the urea group relative to the salicylic acid, which breaks the intramolecular CH⋯O interaction.
It is plausible that gelation by compounds of type 1 and particularly type 2 arises from urea α-tape formation, and the fact that this interaction is much shorter in 2a is consistent with the much more effective gelation behaviour of this compound compared to compounds of type 1. While mono(urea)s interact with one another through a single bifurcated acceptor R12(6) synthon, covalently bound bis(urea)s enjoy a multivalent46 stabilization by virtue of the double R12(6) interaction formed by each molecule. In the present case we postulate that the strong non-covalent dimerization arising from the salicylic acid groups in compounds of type 1 and 2 allows them to behave as pseudo-bis(urea)s with the urea groups in hydrogen bonded pairs held in a centrosymmetric and hence antiparallel arrangement by the centrosymmetric R22(8) carboxylic acid dimer arrangement. Evidence for the persistence of the dimeric structure in the gas phase comes from the ES+ mass spectrum of 2a which, in addition to the molecular ion peak at m/z 252.8, exhibits a distinct peak at 505.1 of approximately half the intensity of the molecular ion, assigned to the dimer. Similar gas phase dimers are readily apparent in the ES+-MS spectra of all compounds of type 1 and 2. In solution the 1H NMR spectrum of 2a in DMSO-d6 solution did not exhibit any change in the chemical shift of the carboxylic acid OH resonance as a function of temperature in the range 20–100 °C, nor in the concentration range 1–8% w/v. In contrast, a modest chemical shift change was observed for the NH resonances, suggesting that while aggregation by urea–urea interaction is in equilibrium under the conditions used in the NMR experiment, the carboxylic acid groups remain fully dimerised under the conditions studied even in a competitive solvent such as DMSO. It is also possible that the lack of chemical shift change as a function of concentration and temperature indicates no interaction at all and the molecule is monomeric; however, this seems unlikely given the strong hydrogen bonding in the salicylic acid group, the ES+-MS evidence and the contrasting behaviour of the generally more weakly hydrogen bonded urea group.47 Definitive proof for the retention of the dimeric structure in solution was obtained from IR spectroscopy. The solid-state ATR spectra of all compounds of type 1 and 2 closely resembled the spectra in the non-competitive solvent 1,2-dibromoethane with a single low frequency asymmetric carbonyl stretching band at 1660 cm−1 for 2a, for example, assigned to the carboxylic acid dimer observed in the X-ray crystal structures. In the more competitive DMSO solution, however, two carboxylic acid carbonyl bands appeared, at 1703 and 1655 cm−1 assigned to monomeric and dimeric carboxylic acids respectively. Other consistent changes were observed throughout the spectra. Hence in the competitive solvent the compound exists as a mixture of monomer and dimer while in non-polar media it is fully dimerised.
Evidence for the relevance of the single crystal X-ray structural data to the gels formed by compounds of type 2 was sought from X-ray powder diffraction.48–50 However the XRPD patterns of the xerogels of 2a, 2c and 2d from nitromethane and nitrobenzene all showed the materials to be amorphous. Given the simplicity of the compounds and the consistent structural results, however, it remains a plausible working hypothesis that the local structure of the gel fibres is closely related to the interactions found in the single crystals.
Given that both the 4-ASA derivatives of type 1 and 5-ASA analogues of type 2 share a common dimeric structure, it is interesting to see that compounds of type 2 exhibit shorter urea tape hydrogen bonding and are also significantly more effective gelators of the solvents studied than compounds of type 1. The out-of-plane twist of the urea group compared to the aromatic ring plane in 2a is exemplified by a torsion angle of 50.9°, significantly larger than those of the 4-ASA derivatives which are between 17.3–38.0°. Gas-phase DFT calculations suggest that this is not solely a crystal packing effect, with the rotation energy barrier for the 4-ASA derivatives being around 29 kJ mol−1, nearly double the analogous barrier for the 5-ASA analogues (Fig. S6, ESI†). This more restricted rotation is likely to be due to the greater conjugation of the urea nitrogen atom in the 4-ASA derivative (Fig. S8, ESI†) as a result of the para-electron withdrawing effect of the carboxylic acid functionality.51 As a result the hydrogen bond acceptor ability of the urea carbonyl group is reduced for steric reasons and hence its ability to express the gel-forming urea α-tape motif is limited.45,52 Overall, the short NH⋯O interactions in X-ray structure of 2a indicate that the urea α-tape is significantly stronger than those displayed by the 4-ASA analogues of type 1. Interestingly, this electron withdrawing effect is evident in the structure of 4-ASA itself which is neutral, while 5-ASA is a zwitterion because of the higher amine basicity.37
In conclusion this work has shown that a robust supramolecular linkage can replace a covalently bonded spacer in bis(urea)s, allowing mono(urea)s to act as effective gelators by means of supramolecular multivalency. In addition to the formation of multiple hydrogen bonded chain interactions, gelation is favoured by non-coplanarity of the urea group and the aryl substituent, maximising the exposure and hence hydrogen bond basicity of the urea carbonyl acceptor. The presence of sterically bulky branched substituents as in 2b also apparently reduces gelation efficiency, again likely as a result of reduced steric accessibility of the urea carbonyl group and hence reduced hydrogen bonded chain growth rate.
We thank the EPSRC for support of this work (EP/J013021/1).
Notes and references
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS PubMed.
- J. F. Miravet and B. Escuder, in Supramolecular Systems in Biomedical Fields, ed. H.-J. Schneider, Royal Society of Chemistry, Cambridge, 2013, p. 331 Search PubMed.
- S. Díaz-Oltra, C. Berdugo, J. F. Miravet and B. Escuder, New J. Chem., 2015, 39, 3785 RSC.
- B. Escuder, F. Rodriguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044 RSC.
- Z. Yang, K. Xu, L. Wang, H. Gu, H. Wei, M. Zhang and B. Xu, Chem. Commun., 2005, 4414 RSC.
- J. J. Panda, A. Mishra, A. Basu and V. S. Chauhan, Biomacromolecules, 2008, 9, 2244 CrossRef CAS PubMed.
- S. Bhuniya, Y. J. Seo and B. H. Kim, Tetrahedron Lett., 2006, 47, 7153 CrossRef CAS.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856 RSC.
- B. Adhikari and A. Banerjee, Soft Matter, 2011, 7, 9259 RSC.
- D. K. Smith, in Organic Nanostructures, ed. L. J. Atwood and J. W. Steed, Wiley-VCH, Weinheim, Germany, 2008, p. 111 Search PubMed.
- C. Sudha, P. Parimaladevi and K. Srinivasan, Mater. Sci. Eng., C, 2015, 47, 150 CrossRef CAS PubMed.
- Y. Diao, K. E. Whaley, M. E. Helgeson, M. A. Woldeyes, P. S. Doyle, A. S. Myerson, T. A. Hatton and B. L. Trout, J. Am. Chem. Soc., 2012, 134, 673 CrossRef CAS PubMed.
- D. J. Adams, K. Morris, L. Chen, L. C. Serpell, J. Bacsa and G. M. Day, Soft Matter, 2010, 6, 4144 RSC.
- J. A. Foster, M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037 CrossRef CAS PubMed.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080 RSC.
- A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540 RSC.
- F. Rodríguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478 CrossRef PubMed.
- H. Yang, T. Yi, Z. Zhou, Y. Zhou, J. Wu, M. Xu, F. Li and C. Huang, Langmuir, 2007, 23, 8224 CrossRef CAS PubMed.
- X. Yu, X. Cao, L. Chen, H. Lan, B. Liu and T. Yi, Soft Matter, 2012, 8, 3329 RSC.
- Y. Zhou, M. Xu, T. Yi, S. Xiao, Z. Zhou, F. Li and C. Huang, Langmuir, 2007, 23, 202 CrossRef CAS PubMed.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed.
- K. Araki and I. Yoshikawa, Top. Curr. Chem., 2005, 256, 133 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS PubMed.
- V. K. Potluri and A. D. Hamilton, J. Supramol. Chem., 2002, 2, 321 CrossRef CAS.
- M. George, G. Tan, V. T. John and R. G. Weiss, Chem. – Eur. J., 2005, 11, 3243 CrossRef CAS PubMed.
- C. Wang, D. Zhang and D. Zhu, J. Am. Chem. Soc., 2005, 127, 16372 CrossRef CAS PubMed.
- S.-Y. Hsueh, C.-T. Kuo, T.-W. Lu, C.-C. Lai, Y.-H. Liu, H.-F. Hsu, S.-M. Peng, C.-h. Chen and S.-H. Chiu, Angew. Chem., Int. Ed., 2010, 49, 9170 CrossRef CAS PubMed.
- G. O. Lloyd, M. O. M. Piepenbrock, J. A. Foster, N. Clarke and J. W. Steed, Soft Matter, 2012, 8, 204 RSC.
- C. Deng, R. Fang, Y. Guan, J. Jiang, C. Lin and L. Wang, Chem. Commun., 2012, 48, 7973 RSC.
- M. Takizawa, A. Kimoto and J. Abe, Dyes Pigm., 2011, 89, 254 CrossRef CAS.
- S.-Y. Hsueh, C.-T. Kuo, T.-W. Lu, C.-C. Lai, Y.-H. Liu, H.-F. Hsu, S.-M. Peng, C.-h. Chen and S.-H. Chiu, Angew. Chem., Int. Ed., 2010, 49, 9170 CrossRef CAS PubMed.
- C. Wang, D. Q. Zhang and D. B. Zhu, Langmuir, 2007, 23, 1478 CrossRef CAS PubMed.
- M. de Loos, A. Friggeri, J. van Esch, R. M. Kellogg and B. L. Feringa, Org. Biomol. Chem., 2005, 3, 1631 CAS.
- L. Applegarth, N. Clarke, A. C. Richardson, A. D. M. Parker, I. Radosavljevic-Evans, A. E. Goeta, J. A. K. Howard and J. W. Steed, Chem. Commun., 2005, 5423 RSC.
- F. S. Schoonbeek, J. H. van Esch, R. Hulst, R. M. Kellogg and B. L. Feringa, Chem. – Eur. J., 2000, 6, 2633 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- R. Montis and M. B. Hursthouse, CrystEngComm, 2012, 14, 5242 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Commun., 2008, 2644, 10.1039/b804259d.
- K. Hyun, S. H. Kim, K. H. Ahn and S. J. Lee, J. Non-Newtonian Fluid Mech., 2002, 107, 51 CrossRef CAS.
- K. Hyun, J. Nam, M. Wilhellm, K. Ahn and S. Lee, Rheol. Acta, 2006, 45, 239 CrossRef CAS.
- F. H. Allen, W. D. S. Motherwell, P. R. Raithby, G. P. Shields and R. Taylor, New J. Chem., 1999, 23, 25 RSC.
- M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415 CrossRef CAS.
- L. S. Reddy, S. Basavoju, V. R. Vangala and A. Nangia, Cryst. Growth Des., 2005, 6, 161 Search PubMed.
- J. H. van Esch, S. DeFeyter, R. M. Kellogg, F. DeSchryver and B. L. Feringa, Chem. – Eur. J., 1997, 3, 1238 CrossRef CAS.
- L. Baldini, A. Casnati, F. Sansone and R. Ungaro, Chem. Soc. Rev., 2007, 36, 254 RSC.
- C. Laurence and M. Berthelot, Perspect. Drug Discovery Des., 2000, 18, 39 CrossRef CAS.
- C. Baddeley, Z. Yan, G. King, P. M. Woodward and J. D. Badjić, J. Org. Chem., 2007, 72, 7270 CrossRef CAS PubMed.
- U. K. Das, D. R. Trivedi, N. N. Adarsh and P. Dastidar, J. Org. Chem., 2009, 74, 7111 CrossRef CAS PubMed.
- D. R. Trivedi, A. Ballabh, P. Dastidar and B. Ganguly, Chem. – Eur. J., 2004, 10, 5311 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, California, USA, 2006 Search PubMed.
- J. H. van Esch, F. Schoonbeek, M. de Loos, H. Kooijman, A. L. Spek, R. M. Kellogg and B. L. Feringa, Chem. – Eur. J., 1999, 5, 937 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of gelation experiments and single crystal structure determination in CIF format. CCDC 1419680–1419682. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc06995e. The underlying research data for this paper is available in accordance with EPSRC open data policy from http://10.15128/kk91fm27z |
| This journal is © The Royal Society of Chemistry 2016 |