
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA combination of polyunsaturated fatty acid, nonribosomal peptide and polyketide biosynthetic machinery is used to assemble the zeamine antibiotics†
Joleen
Masschelein
ab,
Charlien
Clauwers
ab,
Ufedo R.
Awodi
c,
Karen
Stalmans
a,
Wesley
Vermaelen
a,
Eveline
Lescrinier
d,
Abram
Aertsen
b,
Chris
Michiels
b,
Gregory L.
Challis
*c and
Rob
Lavigne
*a
aLaboratory of Gene Technology, KU Leuven, Kasteelpark Arenberg 21-box 2462, B-3001 Heverlee, Belgium. E-mail: rob.lavigne@biw.kuleuven.be; Fax: +32 (0) 16 32 19 65; Tel: +32 (0) 16 37 95 24
bLaboratory of Food Microbiology, KU Leuven, Kasteelpark Arenberg 22, B-3001 Heverlee, Belgium
cDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: g.l.challis@warwick.ac.uk; Fax: +44 (0) 2476 524112; Tel: +44 (0) 2476 574024
dLaboratory of Medicinal Chemistry, Rega Institute for Medical Research, KU Leuven, Minderbroedersstraat 10, B-3000 Leuven, Belgium
First published on 15th October 2014
Abstract
The zeamines are a unique group of antibiotics produced by Serratia plymuthica RVH1 that contain variable hybrid peptide–polyketide moieties connected to a common pentaamino-hydroxyalkyl chain. They exhibit potent activity against a broad spectrum of Gram-positive and Gram-negative bacteria. Here we report a combination of targeted gene deletions, high resolution LC-MS(/MS) analyses, in vitro biochemical assays and feeding studies that define the functions of several key zeamine biosynthetic enzymes. The pentaamino-hydroxyalkyl chain is assembled by an iterative multienzyme complex (Zmn10–13) that bears a close resemblance to polyunsaturated fatty acid synthases. Zmn14 was shown to function as an NADH-dependent thioester reductase and is proposed to release a tetraamino-hydroxyalkyl thioester from the acyl carrier protein domain of Zmn10 as an aldehyde. Despite the intrinsic ability of Zmn14 to catalyze further reduction of aldehydes to alcohols, the initially-formed aldehyde intermediate is proposed to undergo preferential transamination to produce zeamine II. In a parallel pathway, hexapeptide-monoketide and hexapeptide-diketide thioesters are generated by a hybrid nonribosomal peptide synthetase-polyketide synthase multienzyme complex (Zmn16–18) and subsequently conjugated to zeamine II by a stand-alone condensing enzyme (Zmn19). Structures for the resulting prezeamines were elucidated using a combination of high resolution LC-MS/MS and 1- and 2-D NMR spectroscopic analyses. The prezeamines are hypothesized to be precursors of the previously-identified zeamines, which are generated by the action of Zmn22, an acylpeptide hydrolase that specifically cleaves the N-terminal pentapeptide of the prezeamines in a post-assembly processing step. Thus, the zeamine antibiotics are assembled by a unique combination of nonribosomal peptide synthetase, type I modular polyketide synthase and polyunsaturated fatty acid synthase-like biosynthetic machinery.
Introduction
To keep up with the increasing emergence of multidrug-resistant bacterial infections, there is an urgent need for the discovery of novel classes of antibiotics. Natural products have been and continue to be the major source of novel drug leads for the treatment of a wide range of infectious diseases.1 Polyketides and nonribosomal peptides, two major classes of bacterial secondary metabolites, constitute an enormous group of complex, biologically active natural products. They are biosynthesized by large enzymatic assembly lines that employ analogous strategies for sequential linking of short carboxylic acid and amino acid building blocks, respectively.2–4 Like nonribosomal peptide synthetases (NRPSs), type I modular polyketide synthases (PKSs) are modular multienzymes. In both cases, each module consists of a set of discrete catalytic domains responsible for the incorporation of one building block and a variety of modifications into the growing chain. Type I iterative PKSs are multienzymes that use a single set of catalytic domains repeatedly to perform several successive condensation and modification reactions.5 A minimal PKS module consists of three domains: an acyl transferase (AT) domain, which selects and loads an acyl thioester building block onto the phosphopantetheinyl arm of an acyl carrier protein (ACP) domain, and a ketosynthase (KS) domain, which catalyzes chain elongation. In NRPSs, equivalent functions are fulfilled by adenylation (A), peptidyl carrier protein (PCP) and condensation (C) domains, respectively. A variety of additional domains can be found that perform modifications of the growing chain e.g. ketoreductase (KR), dehydratase (DH), enoyl reductase (ER) and C-methyltransferase domains in PKSs and epimerization (E) and N-methylation domains in NRPSs. During the assembly process, biosynthetic intermediates remain covalently tethered to the ACP/PCP domains. Following the final chain elongation/modification cycle, the fully-assembled polyketide or peptide chain is released from the assembly line. The most frequently encountered chain release reaction is catalyzed by a C-terminal thioesterase domain, which catalyzes product release by hydrolysis or macrocyclization.6–8 Alternatively, chain termination can be mediated by thioester reductase (TR) domains or C domains. TR domains release the fully-assembled thioester as an aldehyde using NAD(P)H as a cosubstrate,9 whereas C domains catalyze inter- or intramolecular amide or ester bond formation.10–12Secondary lipid biosynthesis involves the production of long-chain fatty acid products with specialized functions that do not play a direct role in the growth and development of the cell under normal laboratory conditions. Secondary lipid synthase genes show high homology to the pfa clusters of genes involved in the de novo synthesis of long-chain polyunsaturated fatty acids (PUFAs) like eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) in marine, psychrophilic bacteria.13 The pfaA-D cluster typically displays a conserved operon organization and encodes an iterative type I fatty acid synthase (FAS)/PKS system.14,15 Secondary lipid synthases have been shown to produce the alkyl chains of heterocyst glycolipids in nitrogen-fixing cyanobacteria as well as the phenolic lipid alkyl chains in cysts of Azotobacter vinelandii. Additionally, numerous orphan gene clusters encoding related FAS/PKS systems with the same conserved multi-domain architecture have been identified in diverse microbial phyla.13
The zeamines are a unique complex of broad spectrum antibiotics produced by Serratia plymuthica RVH1.16–18 Zeamine production is regulated by a LuxI/LuxR-type quorum sensing system and provides the strain with a selective advantage in competitive environments.19,20 The structures of these antibiotics represent a completely new chemical scaffold consisting of an unusual polyamino alcohol chain (zeamine II, 1, Fig. 1), conjugated via an amide bond to a valine-linked polyketide moiety in the case of zeamine (2) and zeamine I (3) (Fig. 2). The valine residue has been hypothesized to be part of a nonribosomal hexapeptide leader sequence that is cleaved during a post-assembly processing step.17 Specific components of the zeamine complex have also been reported to be produced by the bacterial phytopathogens Dickeya zeae DZ121 (zeamine) and D. zeae EC122 (zeamine and zeamine II). Previously, zeamine biosynthesis was mapped to a 50 kb gene cluster in S. plymuthica RVH1, which appears to be conserved across different bacterial species.17,23,24 In addition to presumed tailoring (Zmn15, Zmn22) and export-related (Zmn20–21) enzymes, two separate assembly lines are encoded by genes within the gene cluster: a type I FAS/PKS homologous to PUFA synthases (Zmn10–14) and a hybrid NRPS/PKS (Zmn16–19). This unusual genetic architecture strongly suggests that an unprecedented combination of NRPS, PKS and PUFA synthase-like multienzymes participates in zeamine assembly.17 To validate this hypothesis and experimentally confirm the functional role of specific enzymes in zeamine biosynthesis, in vitro biochemical assays and feeding studies were performed, and targeted in-frame gene deletions were made in the zeamine biosynthetic gene cluster. The effect of knockout mutagenesis on metabolite production was examined by ultra-high resolution LC-MS analysis of culture supernatants and antibacterial activity assays. Furthermore, the prezeamines, precursors to zeamine and zeamine I, were isolated, structurally elucidated and shown to undergo an unusual post-assembly proteolytic processing step.
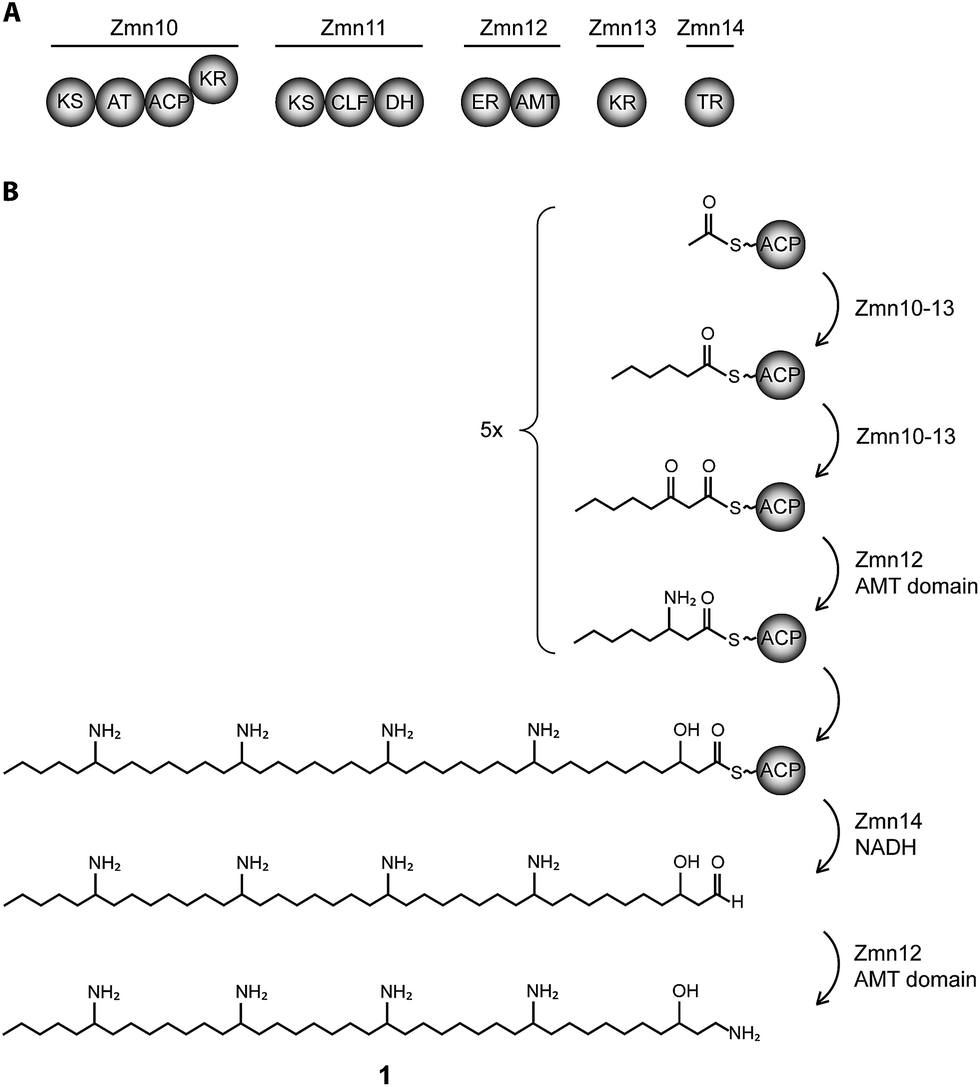 | ||
| Fig. 1 Biosynthesis of zeamine II (1). (A) Domain and module organization of Zmn10–14 which constitute a type I iterative FAS/PKS showing high homology to PUFA synthases. (B) Proposed biosynthetic scheme for zeamine II. Domains are abbreviated as follows: KS = ketosynthase, KR = ketoreductase, ACP = acyl carrier protein, CLF = chain length factor, TR = thioester reductase, DH = dehydratase, ER = enoyl reductase, AMT = aminotransferase. | ||
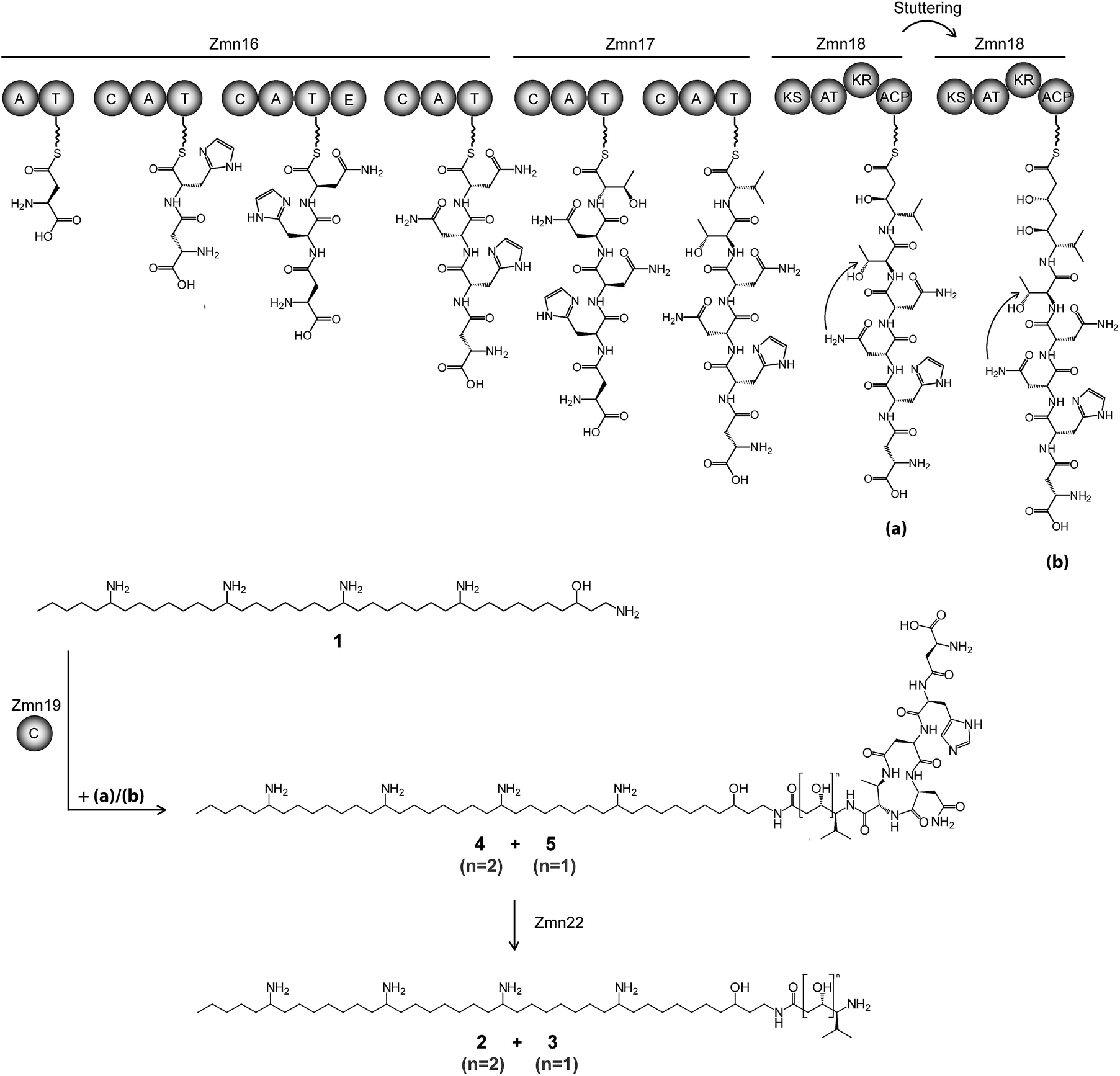 | ||
| Fig. 2 Proposed biosynthetic pathway for zeamine (2) and zeamine I (3). The Zmn16–18 hybrid NRPS/PKS catalyzes the sequential incorporation of γ-Asp, His, Asn, Asn, Thr and Val, followed by one or two elongations of the resulting pentapeptide with a malonyl extender unit. The stand-alone condensation domain Zmn19 condenses the ACP-bound thioesters (a) and (b) to the primary amino group of 1via chain-terminating amide bond formation, giving rise to prezeamine I (5) and prezeamine (4), respectively. Zmn22 then cleaves off the N-terminal pentapeptide in a post-assembly processing step to form 3 and 2. The precise timing of the cyclization reaction between D-Asn and Thr remains unclear. The configurations of the stereocenters depicted here were bioinformatically predicted based on the presence of an epimerization domain in Zmn16 and sequence analysis of the KR domain in Zmn18. Domains are abbreviated as follows: C = condensation, A = adenylation, T = thiolation, KS = ketosynthase, AT = acyltransferase, KR = ketoreductase, ACP = acyl carrier protein. | ||
Results and discussion
Involvement of a PUFA synthase-like multienzyme complex in zeamine II biosynthesis
The 40-carbon pentaamino-alcohol zeamine II (1) is proposed to be biosynthesized by Zmn10–14, an iterative PUFA synthase-like multienzyme complex consisting of three multifunctional proteins and stand-alone ketoreductase (KR) and thioester reductase (TR) enzymes (Fig. 1A). Zmn10–12 show high homology to the PUFA biosynthetic enzymes PfaA, C and D respectively, differing only in the number of ACP and DH domains and in the presence of an unusual PLP-dependent aminotransferase (AMT) domain in Zmn12.17 By analogy to similar domains involved in mycosubtilin and microcystin biosynthesis, this AMT domain likely catalyzes the conversion of selected β-ketothioester intermediates in carbon chain assembly to the corresponding β-aminothioesters, as well as transamination of the aldehyde group in the final tetraamino-alcohol intermediate in zeamine II biosynthesis.25,26 Zmn10 homolog ZmsA (66% amino acid identity) was previously shown to be essential for production of 1 and 2 in D. zeae EC1.22 Antibacterial activity was no longer detectable in mutants in which either zmn10, zmn11, zmn12, zmn13 or zmn14 were individually deleted. LC-MS analysis of culture supernatants from these mutants confirmed that the production of all components of the zeamine complex was abolished (Fig. 3). Moreover, no putative biosynthetic intermediates could be detected. Upon feeding of isolated zeamine II to small-scale cultures of the zmn10, zmn11 and zmn12 deletion mutants, production of 2 and 3 could be observed in the culture supernatant after 72 h fermentation (Fig. S1†). This is consistent with the direct involvement of the PUFA-synthase like multienzyme complex in the biosynthesis of 1. The assembly process is proposed to be initiated by an acetyl-CoA starter unit undergoing two complete rounds of elongation, ketoreduction, dehydration and enoyl reduction (Fig. 1B). Following the next elongation step, the reductive modification of the β-ketothioester is replaced by a transamination, catalyzed by the AMT domain in Zmn12. This process is repeated four more times, with the transamination step being replaced by a ketoreduction in the last iteration. Throughout the biosynthesis, the chain is presumed to be attached to the ACP domain of Zmn10. Reductive release of the fully extended β-hydroxythioester as an aldehyde by Zmn14 and a final transamination reaction would then generate 1. The absolute configurations of the stereocentres bearing the amino groups remain to be elucidated and the enzyme responsible for the introduction of the hydroxyl group into 1 is still to be identified. Comparative sequence analyses previously suggested that the KR domain in Zmn10 would generate a hydroxyl group with an R configuration, whereas the KR domain in Zmn13 would introduce a hydroxyl group with S configuration.17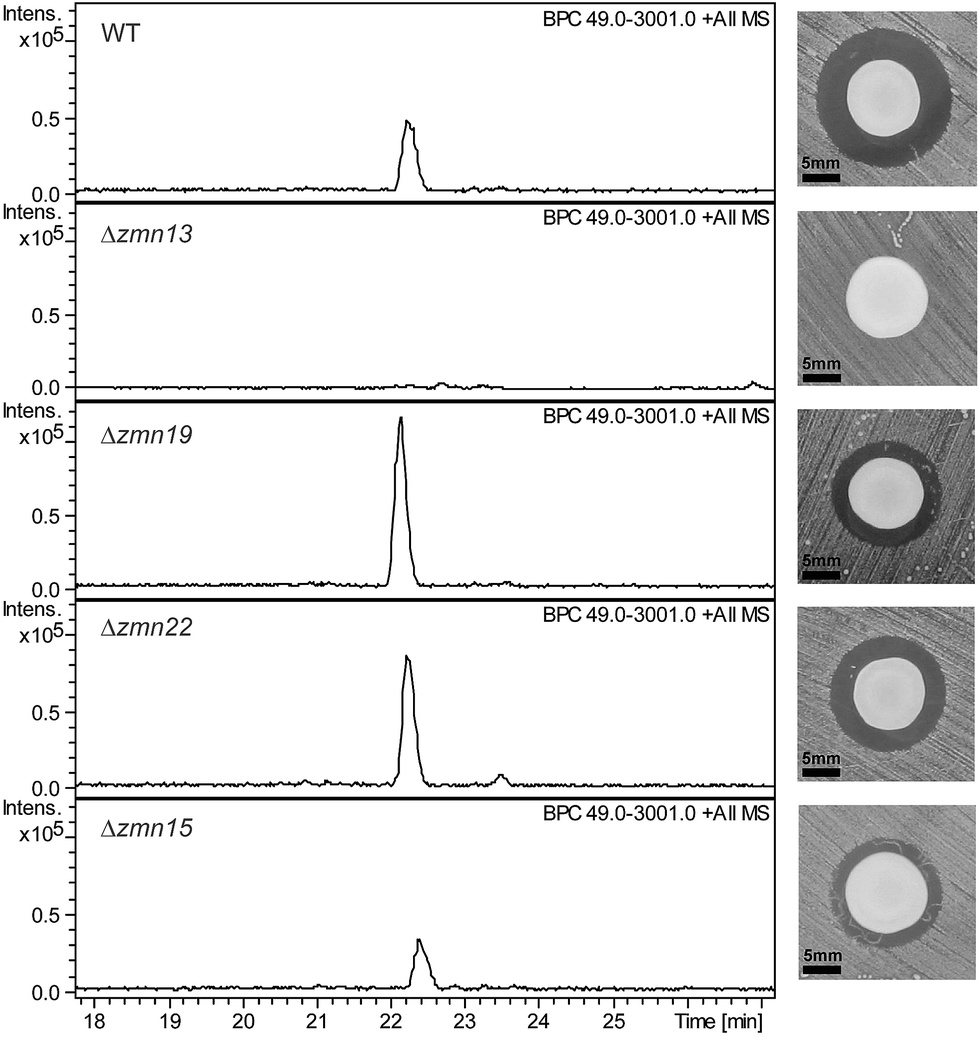 | ||
| Fig. 3 Base peak chromatograms from LC-ESI-MS analyses of culture supernatants from wild type S. plymuthica RVH1 and the deletion mutants. The constituents of the zeamine complex co-elute at 22.2 ± 0.2 minutes. Δzmn22 produces only prezeamines and zeamine II (see Fig. S6†), whereas Δzmn15 produces reduced levels of the zeamine complex. As is the case for Δzmn10, 11, 12 and 14, zeamine production in Δzmn13 cannot be detected. Δzmn19, Δzmn16, Δzmn17 and Δzmn18 produce only zeamine II (see Fig. S5†). The effect of in-frame deletion of the individual zeamine biosynthetic genes on the antimicrobial activity excreted by S. plymuthica RVH1 is shown next to the chromatograms. Plate bioassays were performed using Staphylococcus aureus ATCC27661 as the indicator strain. To rule out the influence of polar effects on the observed phenotypes, deletions were complemented by expressing the respective genes in trans. For an overview of all gene deletion and complementation experiments, see Table S1.† | ||
Zmn14 is a thioester reductase
Zmn14 is proposed to be responsible for reductive release of the tetraamino-hydroxy thioester from the ACP domain of Zmn10 to form the corresponding aldehyde. TR domains typically utilize NAD(P)H to catalyze transfer of a single, or two successive, hydride ions to the carbonyl group of a thioester resulting in formation of an aldehyde or alcohol, respectively.27 To further elucidate the function of Zmn14, it was overproduced in E. coli with a His6-tag fused to its C-terminus and purified to homogeneity using Ni-NTA chromatography. Using dodecanoyl-CoA as a mimic of the natural substrate, the ability of Zmn14 to consume NADPH and NADH was investigated by monitoring the decrease in absorbance at 340 nm (due to production of NAD(P)+). The enzyme was found to have a marked preference for NADH over NADPH and no activity was observed in control reactions utilizing boiled enzyme (Fig. 4). NADH consumption was also observed when Zmn14 was incubated with octanoyl- or butyryl-CoA in place of dodecanoyl-CoA, demonstrating that it is able to tolerate a range of thioester substrates (Fig. S2†). Furthermore, Zmn14 was able to catalyze NADH-dependent reduction of octanal, butanal, 2-octanone and butanone (Fig. S3†). To confirm that such reactions lead to the formation of an alcohol, Zmn14 was incubated with octanal and NADH, and the reaction mixture was extracted with chloroform and treated with N,O-bis-(trimethylsilyl)acetamide (BSA). An analyte with m/z = 187.2, corresponding to the fragment ion of the BSA derivative of octanol resulting from neutral loss of a methyl group, was observed in GC-MS analyses of the extracts (Fig. 5). This analyte was absent from control reactions utilizing boiled enzyme and had the same retention time as the corresponding analyte derived from an authentic standard of the BSA derivative of octanol (Fig. 5).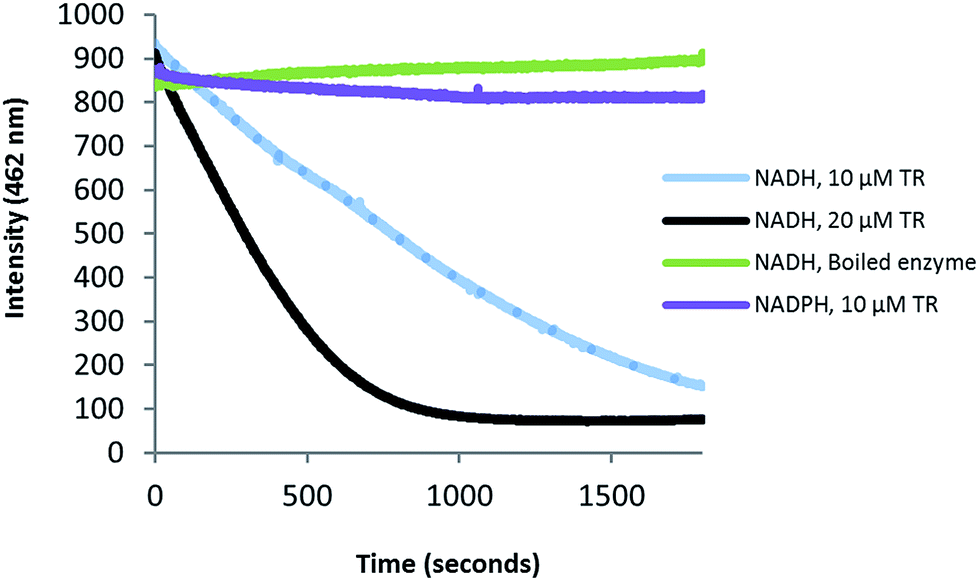 | ||
| Fig. 4 Preferential consumption of NADH over NADPH in the Zmn14-catalyzed reduction of dodecanoyl-CoA. Purified Zmn14 was incubated with dodecanoyl-CoA and either NADH or NADPH and the change in fluorescence emission at 462 nm was monitored. | ||
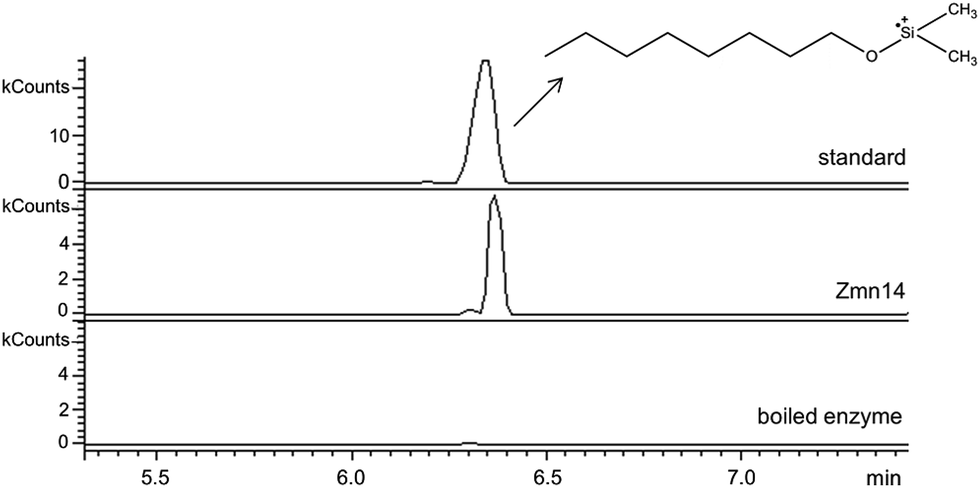 | ||
| Fig. 5 Extracted ion chromatograms at m/z 187.2 from GC-MS analyses of the Zmn14-catalyzed reduction of octanal with NADH. The top chromatogram is for the authentic standard of BSA-derivatized octanol. The middle chromatogram is for the BSA-derivatized product of the enzymatic reaction. The bottom chromatogram corresponds to the BSA-derivatized product of the negative control using boiled enzyme. | ||
Identification of the prezeamines reveals the involvement of a parallel NRPS/PKS assembly line
The valine-derived moieties of zeamine (2) and zeamine I (3) are proposed to originate from the hybrid NRPS/PKS encoded by zmn16–18. This assembly line consists of six NRPS modules and one PKS module (Fig. 2). Although sequence analyses do not indicate that any of the domains are non-functional, only valine, the predicted amino acid substrate of the final NRPS module, is incorporated into the structure of zeamine and zeamine I. This led us to speculate that 2 and 3 arise from ‘prezeamines’ from which an N-terminal pentapeptide is cleaved in a post-assembly processing step.17High resolution LC-MS analyses of wild type S. plymuthica RVH1 culture supernatant identified two metabolites with identical retention times as 1, 2 and 3, and molecular formulae corresponding to zeamine and zeamine I derivatives with five additional amino acids appended to their N-termini (calculated for C69H130N15O12+: 1361.0023, observed: 1361.0009; calculated for C71H134N15O13+: 1405.0286, observed: 1405.0259) (Fig. S4†). Prezeamine (4) was isolated and purified, and its structure was elucidated by a combination of high resolution LC-ESI-MS/MS and various 2-D NMR analyses, including multiplicity-edited HSQC, HSQC-TOCSY and HMBC. These data revealed that the pentapeptide of 4 consists of (from N- to C-terminus): γ-aspartate, histidine, asparagine, asparagine and threonine, with a cyclization occurring between the side chains of the first asparagine and the threonine residue (Fig. 6, S7–S13, Table S2 and S3†). LC-ESI-MS/MS analysis confirmed that prezeamine I (5) harbors the same N-terminal pentapeptide. This peptide moiety is in line with the predicted specificities of the adenylation domains within Zmn16 and Zmn17 and is identical to the proposed peptide part of fabclavine IVa, recently reported as a metabolite of Xenorhabdus szentirmaii (Fig. S14, Table S4†).23 The presence of an epimerization domain in the third NRPS module predicts that the first asparagine residue has the D configuration, in contrast to the five other amino acid residues which are all bioinformatically predicted to have the L configuration. The hexapeptidyl thioester predicted to be assembled by Zmn16 and Zmn17 is proposed to undergo one or two rounds of Zmn18-mediated two-carbon chain elongation, with concomitant reduction of the β-keto groups in the chain elongated intermediates. Sequence comparisons of the KR domain in Zmn18 with KR domains of known stereospecificity from other PKSs, predicts the S configuration for the resulting hydroxyl group(s).17 Zmn19 is then predicted to catalyze condensation of the β-hydroxy and β, δ-dihydroxy thioester products of the PKS with the primary amino group of 1, to form prezeamine I and prezeamine, respectively (Fig. 2). A similar catalytic role has been proposed for Zmn19 homolog ZmsK (48% amino acid identity) in D. zeae EC1 by Zhang and coworkers.28 According to this biosynthetic model, deletion of zmn16, zmn17 or zmn18 should abolish the production of the peptide–polyketide moiety, whereas inactivation of zmn19 would prevent the conjugation of this moiety to 1. The corresponding mutants all displayed reduced antimicrobial activity and LC-MS analyses confirmed that this is due to exclusive production of 1 in all cases (Fig. 3 and S5†), thus providing strong support for our proposed biosynthetic pathway.
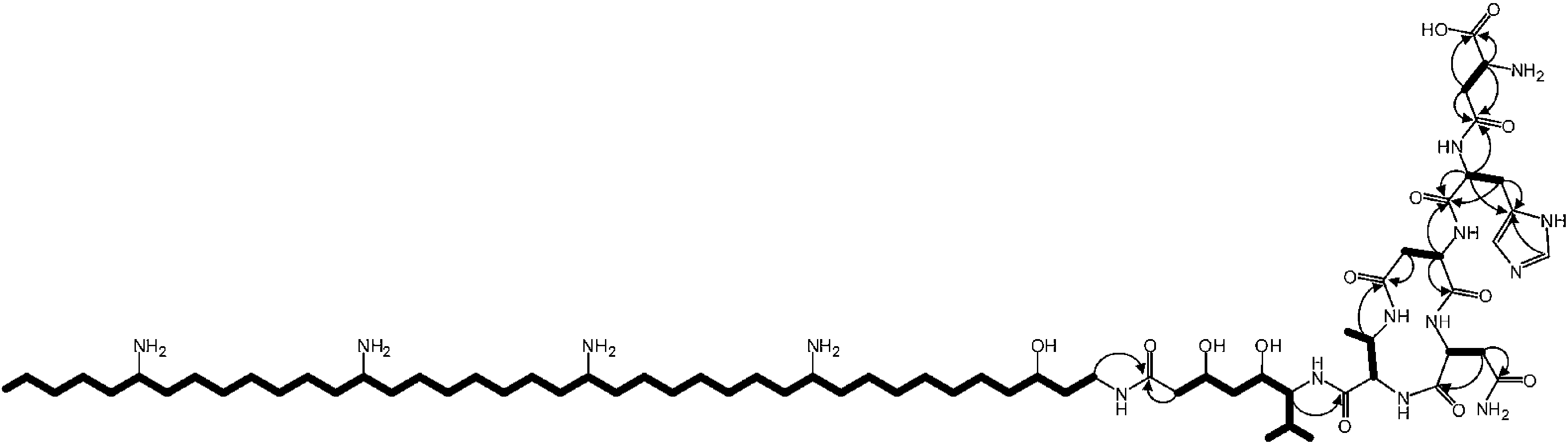 | ||
| Fig. 6 Overview of key 2-D NMR correlations observed for prezeamine (4). Spin systems observed in the 2-D hetero-TOCSY spectrum are depicted as bold lines. Arrows indicate the 1H to quaternary 13C correlations observed in the 2-D HMBC spectrum. | ||
Zmn15, a predicted nitrilase belonging to the C–N hydrolase superfamily of enzymes, which catalyzes condensation and hydrolysis of a wide array of non-peptidic carbon–nitrogen bonds,29 has recently been proposed by Bode and coworkers to catalyze condensation of the threonine and D-asparagine side chains.23 Deletion of zmn15 resulted in reduced production levels of all the zeamines, as indicated by both plate bioassay and LC-MS analysis of culture supernatant (Fig. 3). However, no metabolites with m/z values corresponding to acyclic prezeamines were observed.
Post-assembly processing of the prezeamine precursors
Deletion of zmn22, which encodes a predicted acylpeptide hydrolase, abolished production of 2 and 3, and increased the quantities of 4 and 5 in the culture supernatant relative to the wild type. Zeamine II (1), the metabolic product of Zmn10–14, was also still produced by the zmn22 mutant (Fig. 3, S6†). This is consistent with the involvement of Zmn22 in the conversion of prezeamine (I) to zeamine (I). Although 4 and 5 are overproduced in the zmn22 mutant, a decrease in halo size was observed in the plate bioassay. To better evaluate the relative antibacterial activities, minimal inhibitory concentrations (MIC) were determined for the different zeamine components purified from S. plymuthica RVH1 (Table 1). These experiments demonstrated that the prezeamines are more effective at inhibiting bacterial growth than zeamine II but have a reduced antimicrobial activity compared to the processed zeamines. The cyclic peptide byproduct resulting from prezeamine processing was not detected in cells or culture supernatant from wild type S. plymuthica RVH1. Thus, it remains unclear if this moiety possesses any biological activity.Several products of nonribosomal peptide/polyketide biosynthetic machinery are known to be assembled via inactive precursors that require post-assembly activation to become biologically active. A well-studied example is the biosynthesis of xenocoumacin,30 in which XcnG, a membrane-anchored periplasmic peptidase, specifically cleaves off an N-terminal acylated D-asparagine moiety from the prexenocoumacins upon export to produce bioactive xenocoumacin 1. XcnG homolog ClbP similarly activates precolibactin31 and the same mechanism is proposed to be involved in zwittermycin biosynthesis.32 However, the prezeamine processing mechanism deviates from these other systems. In contrast to XcnG and homologs, no periplasmic signal sequence is present in Zmn22, indicating that peptide cleavage takes place in the cytoplasm. Furthermore, prezeamines are easily detected in supernatants from wild type cultures. This implies that only partial cleavage of prezeamines to zeamines occurs in the cytoplasm and that the prezeamines are exported along with the zeamines as components of a bioactive complex. Interestingly, the gene cluster that directs the biosynthesis of the closely-related fabclavines in Xenorhabdus does not contain a zmn22 homolog and thus only uncleaved metabolites are produced.23 This raises the intriguing question of why a partial pro-peptide cleavage mechanism is operative in the biosynthesis of zeamines by S. plymuthica RVH1.
Conclusions
In summary, we provide evidence for an unprecedented interaction between NRPS, PKS and PUFA synthase-like multienzymes in the biosynthesis of the zeamine antibiotics, an unusual class of polyamine–polyketide–nonribosomal peptide natural products. Using a combination of in vitro biochemical assays, feeding studies and in-frame deletion of biosynthetic genes, the functional roles of most of the enzymes involved in the assembly of the zeamines were experimentally elucidated and insight was obtained into atypical chain release mechanisms from type I PKS and PUFA synthase-like multienzymes. The prezeamines, novel members of the zeamine antibiotic complex containing an N-terminal pentapeptide, were structurally elucidated and implicated as precursors to zeamine and zeamine I, which appear to arise from post-assembly proteolytic processing. This raises intriguing questions regarding the precise biological function of the partial proteolytic processing of the prezeamines in S. plymuthica RVH1.The zeamine biosynthetic pathway illustrates the remarkable level of functional hybridization that can occur between different biosynthetic systems and is a testament of Nature's biosynthetic ingenuity for generating novel natural products with a high degree of structural diversity. This unusual biosynthetic machinery may therefore prove to be a valuable contribution to the synthetic biology toolbox for combinatorial biosynthesis and rational bioengineering to produce novel bioactive compounds. A major focus of future work will be the mechanism of action underlying the broad spectrum of antibacterial activity exhibited by the zeamines.
Acknowledgements
We thank Rebecca Wills, Lijiang Song and Nigam Mishra for assistance with LC-MS(/MS) analyses and Birgit Uytterhoeven for careful reading of the manuscript. The authors acknowledge Sebastien Carpentier for access to the KU Leuven mass spectrometry facility (SyBioMa) and Piet Herdewijn for access to the IVAP lyophilisator. The Bruker MaXis Impact mass spectrometer used in this research was obtained with a grant to GLC from the UK BBSRC (BB/K002341/1). JM is supported by a PhD scholarship from the Research Fund – Flanders (FWO). This study was performed in the framework of the FWO “BaSeics” Research community (W0.014.12N).Notes and references
- J. W. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- S. A. Sieber and M. A. Marahiel, Chem. Rev., 2005, 105, 715–738 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- S. Donadio and L. Katz, Gene, 1992, 111, 51–60 CrossRef CAS.
- P. Caffrey, B. Green, L. C. Packman, B. J. Rawlings, J. Staunton and P. F. Leadlay, Eur. J. Biochem., 1991, 195, 823–830 CrossRef CAS PubMed.
- T. Brautaset, O. N. Sekurova, H. Sletta, T. E. Ellingsen, A. R. Strøm, S. Valla and S. B. Zotchev, Chem. Biol., 2000, 7, 395–403 CrossRef CAS.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- T. A. Keating, G. Marshall and C. T. Walsh, Biochemistry, 2000, 39, 15513–15521 CrossRef CAS PubMed.
- S. Lin, S. G. Van Lanen and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4183–4188 CrossRef CAS PubMed.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS PubMed.
- C. N. Shulse and E. E. Allen, PLoS One, 2011, 6, e20146 CAS.
- H. Okuyama, Y. Orikasa, T. Nishida and N. Morita, Appl. Environ. Microbiol., 2007, 73, 665–670 CrossRef CAS PubMed.
- J. G. Metz, P. Roessler, D. Facciotti, C. Levering, F. Dittrich, M. Lassner, R. Valentine, K. Lardizabal, F. Domergue, A. Yamada, K. Yazawa, V. Knauf and J. Browse, Science, 2001, 293, 290–293 CrossRef CAS PubMed.
- R. Van Houdt, P. Moons, A. Jansen, K. Vanoirbeek and C. W. Michiels, FEMS Microbiol. Lett., 2005, 246, 265–272 CrossRef CAS PubMed.
- J. Masschelein, W. Mattheus, L.-J. Gao, P. Moons, R. Van Houdt, B. Uytterhoeven, C. Lamberigts, E. Lescrinier, J. Rozenski, P. Herdewijn, A. Aertsen, C. Michiels and R. Lavigne, PLoS One, 2013, 8, e54143 CAS.
- R. Van Houdt, D. Van der Lelie, J. A. Izquierdo, A. Aertsen, J. Masschelein, R. Lavigne, C. W. Michiels and S. Taghavi, Genome Announc., 2014, 2(pii), e00021-14 CrossRef PubMed.
- R. Van Houdt, P. Moons, A. Aertsen, A. Jansen, K. Vanoirbeek, M. Daykin, P. Williams and C. W. Michiels, Res. Microbiol., 2007, 158, 150–158 CrossRef CAS PubMed.
- P. Moons, R. Van Houdt, A. Aertsen, K. Vanoirbeek, Y. Engelborghs and C. W. Michiels, Appl. Environ. Microbiol., 2006, 72, 7294–7300 CrossRef CAS PubMed.
- J. Wu, H. B. Zhang, J. L. Xu, R. J. Cox, T. J. Simpson and L. H. Zhang, Chem. Commun., 2010, 46, 333–335 RSC.
- J. Zhou, H. Zhang, J. Wu, Q. Liu, P. Xi, J. Lee, J. Liao, Z. Jiang and L. H. Zhang, Mol. Plant-Microbe Interact., 2011, 24, 1156–1164 CrossRef CAS PubMed.
- S. W. Fuchs, F. Grundmann, M. Kurz, M. Kaiser and H. B. Bode, ChemBioChem, 2014, 15, 512–516 CrossRef CAS PubMed.
- L. Garlant, P. Koskinen, L. Rouhiainen, P. Laine, L. Paulin, P. Auvinen, L. Holm and M. Pirhonen, Diversity, 2013, 5, 824–842 CrossRef PubMed.
- Z. D. Aron, P. C. Dorrestein, J. R. Blackhall, N. L. Kelleher and C. T. Walsh, J. Am. Chem. Soc., 2005, 127, 14986–14987 CrossRef CAS PubMed.
- L. Rouhiainen, T. Vakkilainen, B. L. Siemer, W. Buikema, R. Haselkorn and K. Sivonen, Appl. Environ. Microbiol., 2004, 70, 686–692 CrossRef CAS.
- A. Chhabra, A. S. Haque, R. K. Pal, A. Goyal, R. Rai, S. Joshi, S. Panjikar, S. Pasha, R. Sankaranarayanan and R. S. Gokhale, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5681–5686 CrossRef CAS PubMed.
- Y. Cheng, X. Liu, S. An, S. Chang, Y. Zou, L. Huang, J. Zhong, Q. Liu, Z. Jiang, J. Zhou and L. H. Zhang, Mol. Plant-Microbe Interact., 2013, 26, 1294–1301 CrossRef CAS PubMed.
- H. C. Pace and C. Brenner, Genome Biol., 2001, 2, 0001.1–0001.9 CrossRef.
- D. Reimer, K. M. Pos, M. Thines, P. Grün and H. B. Bode, Nat. Chem. Biol., 2011, 7, 888–890 CrossRef CAS PubMed.
- C. A. Brotherton and E. P. Balskus, J. Am. Chem. Soc., 2013, 135, 3359–3362 CrossRef CAS PubMed.
- B. M. Kevany, D. A. Rasko and M. G. Thomas, Appl. Environ. Microbiol., 2009, 75, 1144–1155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete description of experimental details, and additional tables, figures, chromatograms, spectra, results from biological assays and LC-MS(/MS)- and NMR data for the prezeamines. See DOI: 10.1039/c4sc01927j |
| This journal is © The Royal Society of Chemistry 2015 |