
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tandem redox mediator/Ni(II) trihalide complex photocycle for hydrogen evolution from HCl†
Seung Jun
Hwang
a,
David C.
Powers
a,
Andrew G.
Maher
ab and
Daniel G.
Nocera
*a
aDepartment of Chemistry and Chemical Biology, 12 Oxford Street, Cambridge, MA 02138-2902, USA. E-mail: dnocera@fas.harvard.edu
bDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139-4307, USA
First published on 8th October 2014
Abstract
Photoactivation of M–X bonds is a challenge for photochemical HX splitting, particularly with first-row transition metal complexes because of short intrinsic excited state lifetimes. Herein, we report a tandem H2 photocycle based on combination of a non-basic photoredox phosphine mediator and nickel metal catalyst. Synthetic studies and time-resolved photochemical studies have revealed that phosphines serve as photochemical H-atom donors to Ni(II) trihalide complexes to deliver a Ni(I) centre. The H2 evolution catalytic cycle is closed by sequential disproportionation of Ni(I) to afford Ni(0) and Ni(II) and protolytic H2 evolution from the Ni(0) intermediate. The results of these investigations suggest that H2 photogeneration proceeds by two sequential catalytic cycles: a photoredox cycle catalyzed by phosphines and an H2-evolution cycle catalyzed by Ni complexes to circumvent challenges of photochemistry with first-row transition metal complexes.
Introduction
The photochemical splitting of hydrohalic acids (HX) into H2 and X2 is an approach to solar fuel synthesis1 that stores a comparable amount of energy to water splitting. In addition to the similar energy densities implicit in HX and H2O splitting chemistries, HX splitting mandates management of only two electrons and two protons, whereas H2O splitting requires management of four protons and four electrons.2–5 Photocatalytic HX splitting requires accomplishing multielectron photochemical reactions to activate strong M–X bonds. Typically, photoreduction has been the limiting step in HX splitting photocatalysis and often X2 elimination requires the use of chemical traps for evolved halogen equivalents.6–17 Attempts at promoting HX splitting with first-row transition metal complexes are attractive given that these metals are typically earth abundant, but have been largely unsuccessful. The challenge of using first-row metal complexes as HX splitting photocatalysts is that photochemical activation of Ni(I) or Ni(II) halides frequently does not lead to photoreduction reactions,18,19 likely due to the short excited state lifetimes of first-row transition metal complexes.20–22To overcome the short excited state lifetimes typical of first-row complexes, we have pursued a photoredox strategy for H2 evolution from HCl in which the photochemistry and H2 evolution roles are separated between a photoredox mediator and a hydrogen-evolution catalyst, respectively.23 We were attracted to this strategy because it does not rely on molecular excited states of first-row metal complexes. In our first foray into photoredox catalysis for H2 evolution, we employed bipyridines as photoredox mediators and Ni polypyridyl complexes as H2 evolution catalysts. H-atom abstraction (HAA) by the excited state of the bipyridine afforded a pyridinyl radical, which engaged with a Ni(II) halide complex to generate a Ni(I) intermediate via halogen radical abstraction. The resulting Ni(I) complex underwent disproportionation to a Ni(II) complex and a Ni(0) species, which subsequently engaged in protolytic H2 evolution. While these efforts demonstrated a synthetic cycle for H2 evolution, H2-evolution catalysis was not observed because the basic bipyridyl photoredox mediator was passivated in the presence of HCl.
To address the challenges of photoredox catalysis for H2 evolution from HCl, we turned our attention to identifying a photoredox mediator that could function under acidic conditions. We now examine the role of phosphines as photoredox mediators under acidic conditions (pKa in CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PPh3 = 7.64; pyridine = 12.53).24 The photochemical homolysis of P–H bonds of 2° phosphines generates phosphinyl radicals that display sufficient lifetime (∼160 μs) to participate in halogen-atom abstraction from a Ni(II) halide complex to furnish a reduced Ni intermediate that participates in an H2 evolution cycle; the phosphine photoredox mediator is regenerated by HAA from solvent to close the photocycle.25–27 The H2-evolution cycle may eventually be closed by thermally promoted protolytic H2 evolution with HCl.
:PPh3 = 7.64; pyridine = 12.53).24 The photochemical homolysis of P–H bonds of 2° phosphines generates phosphinyl radicals that display sufficient lifetime (∼160 μs) to participate in halogen-atom abstraction from a Ni(II) halide complex to furnish a reduced Ni intermediate that participates in an H2 evolution cycle; the phosphine photoredox mediator is regenerated by HAA from solvent to close the photocycle.25–27 The H2-evolution cycle may eventually be closed by thermally promoted protolytic H2 evolution with HCl.
Experimental
Materials and methods
All reactions were carried out in an N2-filled glovebox. Anhydrous solvents were obtained by filtration through drying columns.28 NMR chemical shifts are reported in ppm with the residual solvent resonance as internal standard. UV–vis spectra were recorded at 293 K in quartz cuvettes on a Spectral Instruments 400 series diode array and were blanked against the appropriate solvent. PhICl229 and Ni(PPh3)2(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)30 were prepared according to reported procedures. NiCl2dme (dme = 1,2-dimethoxyethane) and AgOTf (OTf = trifluoromethanesulfonate) were obtained from Strem Chemicals. Cl2PPh3, prepared by treatment of PPh3 with PhICl2, displayed spectral features identical to those reported in the literature.31 NiCl2(PPh3)2 (1), Ni(PPh3)4 (5), tetrabutylammonium chloride (nBu4NCl), tetraethylammonium chloride (nEt4NCl), and triphenylphosphine (PPh3) were obtained from Sigma Aldrich. All chemicals were used without further purification. Elemental analysis was obtained by Complete Analysis Laboratories, Inc., New Jersey. Evolved hydrogen was quantified by gas chromatography using a calibration curve derived from adding HCl to known quantities of NaH; over the relevant concentration range, the gas chromatograph response was linear. This procedure has previously been validated by comparison with Toepler pump combustion analysis.15
CH2)30 were prepared according to reported procedures. NiCl2dme (dme = 1,2-dimethoxyethane) and AgOTf (OTf = trifluoromethanesulfonate) were obtained from Strem Chemicals. Cl2PPh3, prepared by treatment of PPh3 with PhICl2, displayed spectral features identical to those reported in the literature.31 NiCl2(PPh3)2 (1), Ni(PPh3)4 (5), tetrabutylammonium chloride (nBu4NCl), tetraethylammonium chloride (nEt4NCl), and triphenylphosphine (PPh3) were obtained from Sigma Aldrich. All chemicals were used without further purification. Elemental analysis was obtained by Complete Analysis Laboratories, Inc., New Jersey. Evolved hydrogen was quantified by gas chromatography using a calibration curve derived from adding HCl to known quantities of NaH; over the relevant concentration range, the gas chromatograph response was linear. This procedure has previously been validated by comparison with Toepler pump combustion analysis.15
Preparation of Ni(II) trihalide complexes
Preparation of [ClPPh3]OTf
To a solution of Cl2PPh3 (127 mg, 3.81 × 10−4 mol, 1.00 equiv.) in CH2Cl2 was added AgOTf (98.0 mg, 3.81 × 10−4 mol, 1.00 equiv.) as a suspension in CH2Cl2. White solid immediately precipitated when AgOTf was added and the reaction mixture was stirred at 23 °C for 1 h before being filtered through Celite. The filtrate was concentrated in vacuo and the residue was taken up in THF and solvent was decanted, and the residue was dried in vacuo to afford 157 mg of the title compound as a white solid (92% yield). 31P NMR (160 MHz, CD2Cl2) δ (ppm): 66.4; 19F NMR (275 MHz, CD2Cl2) δ (ppm): −78.9. The spectral data is consistent with that reported for ClPPh3·AlCl4.33 Crystals suitable for single-crystal diffraction analysis were obtained from a CH2Cl2 solution layered with Et2O.Preparation of Ni(II) tetrachloride complex [NiCl4][Et4N]2
A solution of nEt4Cl (30.2 mg, 1.82 × 10−4 mol, 1.00 equiv.) in 2 mL of CH2Cl2 was added to a solution of NiCl2(dme) (40.0 mg, 1.82 × 10−4 mol, 1.00 equiv.) in 2 mL of CH2Cl2 to prompt an immediate colour change from yellow to green. After stirring at 23 °C for 0.5 h, the reaction mixture was concentrated to dryness and the residue was taken up in pentane and solvent was decanted, and the residue was dried in vacuo to afford 77.2 mg of the title compound as a green solid (92% yield). Crystals suitable for single-crystal diffraction analysis were obtained from a CH3CN solution layered with Et2O and unit cell data matched literature reports.34Preparation of Ni(I) complexes
CH2) (20.0 mg, 3.30 × 10−5 mol, 1.00 equiv.) and NiCl2(PPh3)2 (21.4 mg, 3.30 × 10−5 mol, 1.00 equiv.) as solids followed by 4 mL of Et2O. The reaction mixture was stirred at 23 °C for 0.5 h, during which time a yellow precipitate was observed. The mixture was concentrated to dryness and the residue was taken up in pentane and dried in vacuo to afford 18.4 mg of the title complex as a yellow solid (90% yield).36
Results and discussion
We targeted phosphines as potential photoredox mediators for a tandem photoredox/transition metal catalysed H2-evolution photocycle from HCl based on their demonstrated ability to serve as photochemical H-atom donors.27 To evaluate the viability of the proposed phosphine-mediated photoredox approach for H2 evolution, we photolyzed Ni(II) complex NiCl2(PPh3)2 (1) in THF (λ > 295 nm) in the presence of 1.0 equiv. PPh3 and 15 equiv. of HCl. The light beige reaction solution turned pale blue upon photolysis. Analysis of the headspace by gas chromatography (GC) confirmed H2 as the exclusive gaseous product under these conditions; integration of the chromatogram and comparison to a H2 calibration curve generated from the reaction of NaH with HCl revealed that 3.1 turnovers had been achieved in 18 h. NiCl2(PPh3)2 participates in ligand dissociation equilibria to release PPh3 (vide infra),37 and thus H2-evolving photocatalysis was also observed in the absence of exogenous PPh3. Evaluation of the amount of H2 evolved as a function of time (Fig. 1), showed that in the presence of excess HCl, H2 evolution continues and 9 turnovers were achieved after 44 h with no signs of deactivation.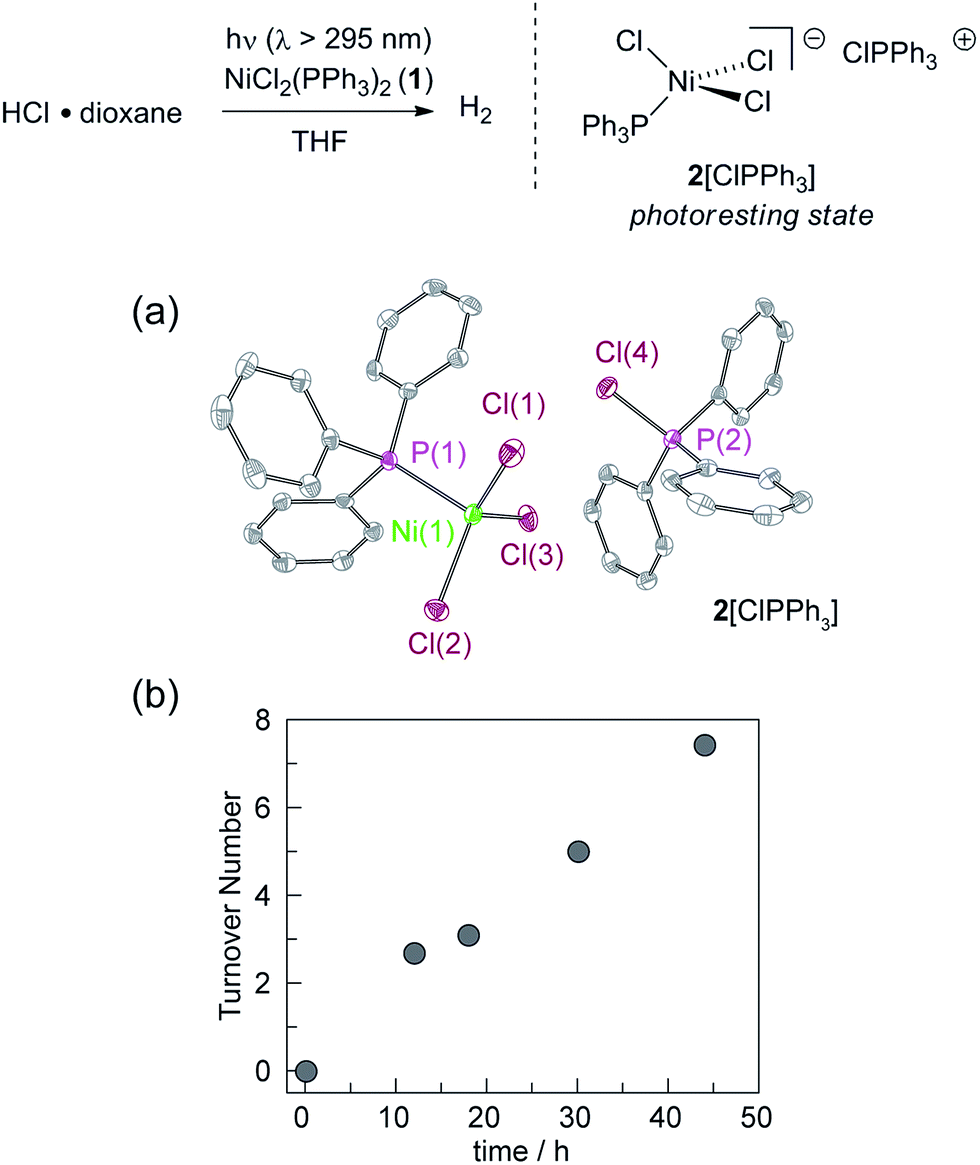 | ||
| Fig. 1 Photolysis of NiCl2(PPh3)2 in THF (λ > 295 nm) in the presence of 15 equiv. of HCl affords H2 as well as 2[ClPPh3]. (a) Thermal ellipsoid plot of 2[ClPPh3] drawn at the 50% probability level. The hydrogen atoms are omitted for clarity. (b) Time-dependent turnover number (TON) of H2 produced by a 4.5 mM THF solution of NiCl2(PPh3)2 in the presence of 15 equiv. HCl (λ > 295 nm). | ||
During the photolysis of NiCl2(PPh3)2, the blue-coloured solution that initially develops persists throughout subsequent H2 evolution and based on UV-vis measurements, appears to represent the photoresting state of the catalyst system. The pale blue photoproduct was identified as Ni(II) trihalide complex 2[ClPPh3] by single-crystal X-ray diffraction analysis.‡ In addition, comparison of the in situ UV-vis spectrum obtained during H2-evolving catalysis with a spectrum obtained from an authentic sample of 2[ClPPh3], prepared by treatment of NiCl2(PPh3)2 with 1.0 equiv. of PhICl2 (Fig. S15†), confirmed the identity of the catalyst resting state. Independently isolated complex 2[ClPPh3] is chemically competent at H2 generation under above photoreaction conditions.
Catalyst resting state 2[ClPPh3] is constituted of a Ni trichloride anion and a phosphonium cation. In order to establish the roles of both the Ni complex and phosphine in the observed H2 evolution reaction, 2[TBA] and phosphonium cation [ClPPh3]OTf were independently prepared. As summarized in Table 1, Ni(II) complex 2[TBA] showed a similar activity toward HCl with 5.0 TON in 18 h. Complex 2[TBA] also participates in a minor equilibrium with free PPh3 (vide infra) and thus both catalyst and photoredox mediator are present when 2[TBA] is employed as the photocatalyst. In contrast, phosphonium salt [ClPPh3]OTf does not produce H2 under the same reaction conditions. Additionally, neither PPh3 or [NiCl4][TEA]2 is a competent H2-evolution catalyst, confirming the necessity of both Ni complex and phosphine for productive H2 evolution chemistry.
|
|
|
|---|---|
| Compound | TON |
| 2[ClPPh3] | 2.0 |
| PPh3 | 0 |
| [NiCl4][Et4N]2 | 0 |
| 2[TBA] | 5.0 |
| [ClPPh3]OTf | 0 |
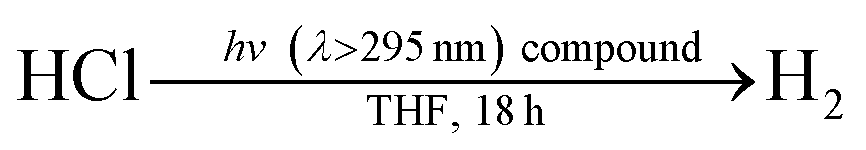
Fig. 2 illustrates a tandem catalytic cycle that accounts for the photogeneration of H2 from HCl catalyzed by the Ni phosphine complexes and phosphine photoredox mediators. Diphenyl phosphine is initially formed by photochemical cleavage of the P–C bond and H-atom abstraction from solvent.27 Photochemical cleavage of the P–H bond in HPPh2 generates an H-atom equivalent and a diphenylphosphinyl radical.27 The H-atom participates in halogen-atom abstraction with Ni(II) resting state 2 to generate a Ni(I) intermediate while the accompanying diphenylphosphinyl radical participates in C–H abstraction with solvent to regenerate the diphenylphosphine and close the photoredox cycle. Ni(I) intermediate 3 undergoes disproportionation to afford NiCl2(PPh3)2 (1) and Ni(PPh3)4 (5). Protonolysis of Ni(0) complex 5 affords H2 and regenerates Ni(II) dihalide 1, thus closing the hydrogen evolution cycle. The number of phosphine ligands bound to intermediates in the tandem cycle illustrated in Fig. 2 is unknown. Isolated complexes 2[ClPPh3], 3, 4 and 5, which display 1–4 phosphine ligands per Ni, are all competent catalysts for H2 evolution and proceed with the same photoresting state (2[ClPPh3]), demonstrating ligand dissociation equilibria37 are established during catalysis (Fig. S16†).
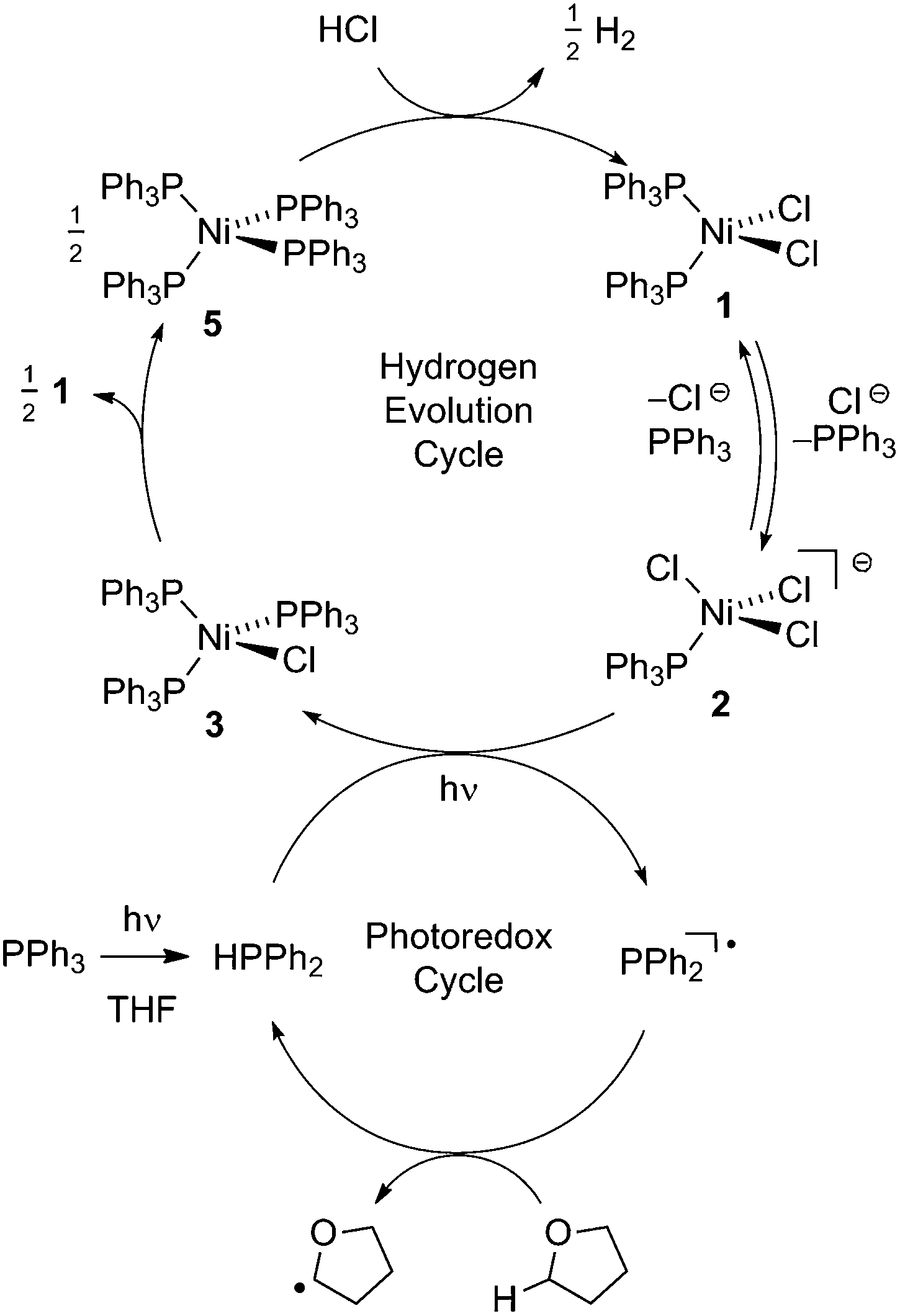 | ||
| Fig. 2 Proposed tandem catalytic cycles for H2-generation with Ni-based H2-evolution catalysts and phosphine-based photoredox mediators. | ||
In order to probe the contention that photogenerated diphenylphosphinyl radicals could mediate the reduction of a Ni–Cl bond from the Ni(II) trihalide complex, we carried out time-resolved photochemical experiments. On the picosecond timescale, a transient absorption (TA) difference spectrum obtained by laser flash photolysis (λexc = 310 nm, THF solutions) of 2[TBA] exhibits a spectral growth centred at 506 nm with a lifetime of ∼700 ps (Fig. S18†). An identical spectral feature was observed during laser flash photolysis of PPh3 solutions. This feature was assigned to be that of the singlet excited state of PPh3, and the observation of this signal in the TA spectrum of 2[TBA] supports the presence of a minor equilibrium between 2[TBA] and free PPh3.38 Additional support for this ligand dissociation equilibrium is the observation that both 2[TBA] and PPh3 both show an emission band centred at 500 nm with a 900 ps lifetime (Fig. S17†), which is well-matched to reported PPh3 photophysics.39 The similar emission lifetimes for both 2[TBA] and PPh3 excludes dynamic quenching of the excited 1PPh3* species by the Ni complex and suggests that the relatively low steady-state emission intensity observed for 2[TBA] is due only to a low equilibrium concentration of PPh3.
The photochemistry of 2 and PPh3 were also examined at longer time scales by nanosecond flash photolysis (Fig. 3). Flash photolysis of either 2[TEA] (red spectrum, Fig. 3) or PPh3 (black spectrum, Fig. 3) leads to the observation of TA signals that are ascribed to diphenylphosphinyl radical.27 The lifetime of the diphenylphosphinyl radical derived from PPh3 with and without the presence of 2[TBA] in solution is the same, consistent with no direct reaction between Ni(II) complex and diphenylphosphinyl radical (Fig. S19†). Substantial phosphine consumption is not required for H2 evolution because the diphenylphosphine generated during catalysis is a competent photoredox carrier. Nanosecond-resolved TA spectra, collected by laser flash photolysis of diphenylphosphine in THF, display the spectral features of diphenylphosphinyl radical (Fig. S20†), confirming that phosphine mediators can be catalytic.27,40
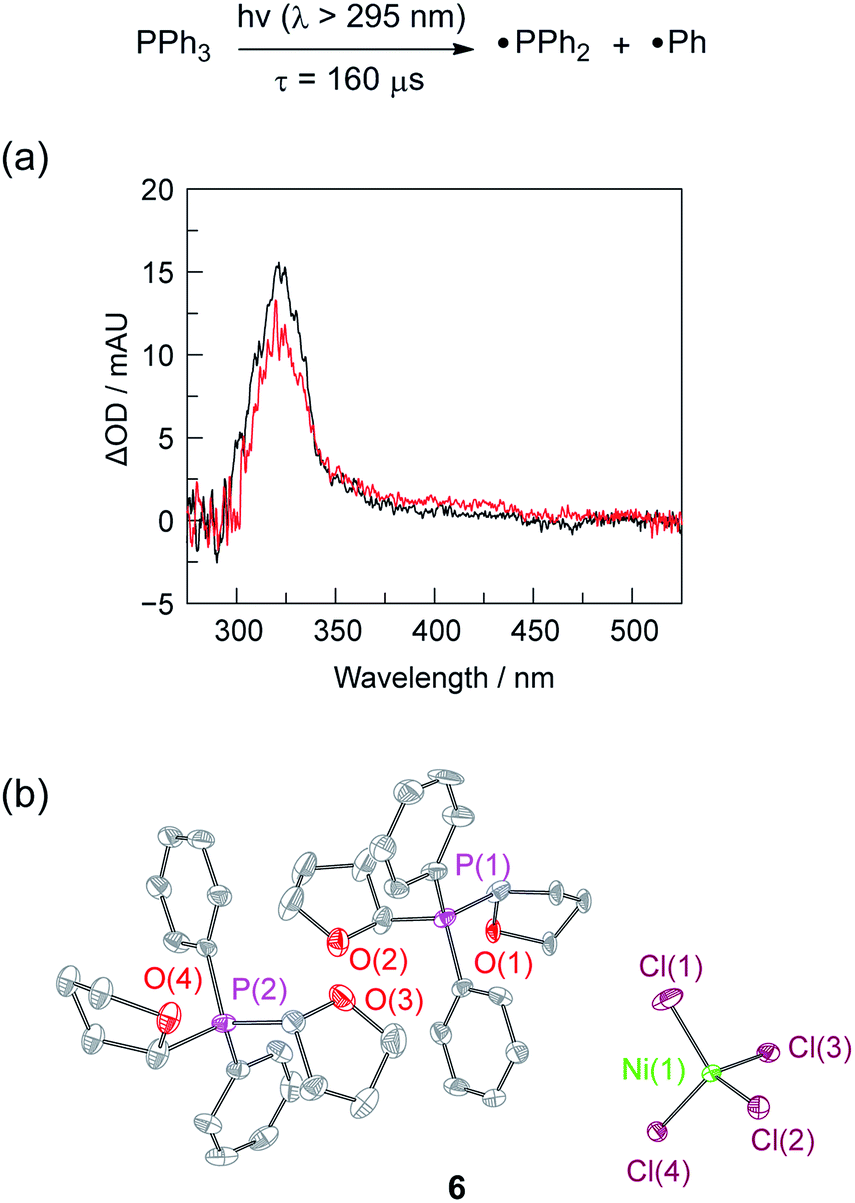 | ||
Fig. 3 (a) Nanosecond transient absorption (TA) spectroscopy of Ni complex 2[TEA] ( , red) and PPh3 ( , red) and PPh3 ( , black) is consistent with formation of diphenylphosphinyl radical. TA spectrum obtained by laser flash photolysis (310 nm pump) of a 1:1 THF:CH3CN solution recorded at a 1 μs delay. (b) Thermal ellipsoid of 6 drawn at the 50% probability level. The hydrogen atoms are omitted for clarity. , black) is consistent with formation of diphenylphosphinyl radical. TA spectrum obtained by laser flash photolysis (310 nm pump) of a 1:1 THF:CH3CN solution recorded at a 1 μs delay. (b) Thermal ellipsoid of 6 drawn at the 50% probability level. The hydrogen atoms are omitted for clarity. | ||
That the photogenerated diphenylphosphinyl radical engages in HAA with THF to produce diphenylphosphine is confirmed by the isolation of complex 6, as a photo-byproduct of the photolysis of Ni complex 2[TEA]. Complex 6 features two C-bound tetrahydrofuranyl ligands on a phosphorus centre and could be derived from reaction of photogenerated diphenylphosphinyl radicals with furanyl radicals derived from HAA chemistry. Additionally we observed (GC-MS) octahydro-2,2′-bifuran (homocoupled THF) as a photochemical byproduct. While HAA from THF by diphenylphosphinyl radicals is endothermic (16 kcal mol−1 uphill based on P–H and C–H bond dissociation energies),41 irreversible subsequent reactions, such as radical coupling to afford homocoupled THF sequester reactive radical intermediates.40
The rate for HAA from solvent by photogenerated diphenylphosphinyl is strongly correlated with solvent C–H bond energies.27,42 We therefore anticipated that efficiency of the total catalytic process would be dictated by the turnover frequency of the photoredox mediator, which depends on the C–H BDEs of H-atom donor. The turnover frequency (TOF) of hydrogen generation from HCl with Ni complex 2 strongly depends on the solvent we employed, showing a positive correlation to the BDE of solvents: 0.34 h−1 TOF in THF, 0.05 h−1 TOF in CH3CN, and 0.02 h−1 TOF in C6H6 (92, 96, and 112 kcal mol−1 of C–H BDEs respectively) (see Fig. S21† for details).43–45
To probe the potential fate of potential Ni(I) intermediates during catalysis, we examined the chemistry of isolated Ni(I) complexes. Ni(I) complex 3 can be isolated from the comproportionation reaction of NiCl2(PPh)2 with Ni(PPh3)4. Based on the E°(NiII/NiI) and E°(NiI/Ni0) measured by cyclic voltammetry in THF (Fig. S22†), comproportionation is thermodynamically favored. In contrast, in the presence of exogenous chloride ion, added as tetrabutylammonium chloride, disproportionation of Ni(I) complex 3 to Ni(II) complex 1 and Ni(0) complex 5 is observed, as determined by both 31P NMR and electronic absorption spectroscopy (Fig. S8 and S14,† respectively). During H2 evolution photocatalysis, chloride is present in large excess (68 mM) with respect to potential Ni(I) intermediates.
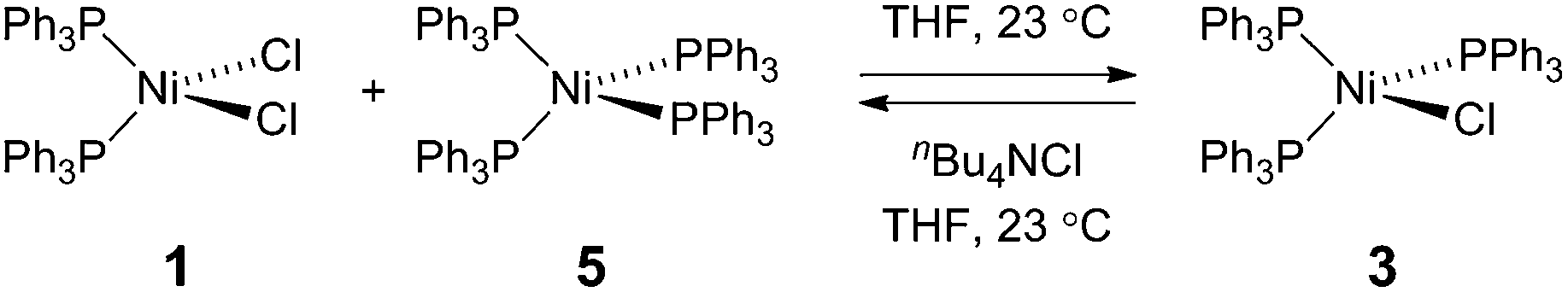 | (1) |
To assess whether the initially produced Ni(I) complex (3) or the Ni(0) complex (5) generated by disproportionation are active for H2 production, we examined the stoichiometric H2-evolution reaction chemistry of Ni(I) and Ni(0) complexes with HCl. The results of these experiments are summarized in Fig. 4. Treatment of Ni complexes 3, 4, and 5 with 15 equiv. HCl in THF generates H2 in 44, 33, and 89% yields, respectively, along with Ni(II) complex 1, NiCl2(PPh3)2. To gain insight into whether H2 evolution proceeds by protonation of Ni(I) or Ni(0), generated by disproportionation reactions, electrochemical H2 evolution was examined using Ni(II) trihalide complex 2[TEA]. As illustrated in Fig. 4, Ni complex 2[TEA] exhibits two electrochemically irreversible waves for the NiII/I and NiI/0 couples at −1.62 and −1.95 V vs. Fc+/Fc, respectively. In the presence of excess HCl (pKa = 8.9 in CH3CN)46 these two peaks exhibit catalytic cathodic waves. The dominant CV features of the NiI/0 wave in the presence of excess HCl are consistent with Ni0 being involved in the H2 generating steps in our photocatalysis.
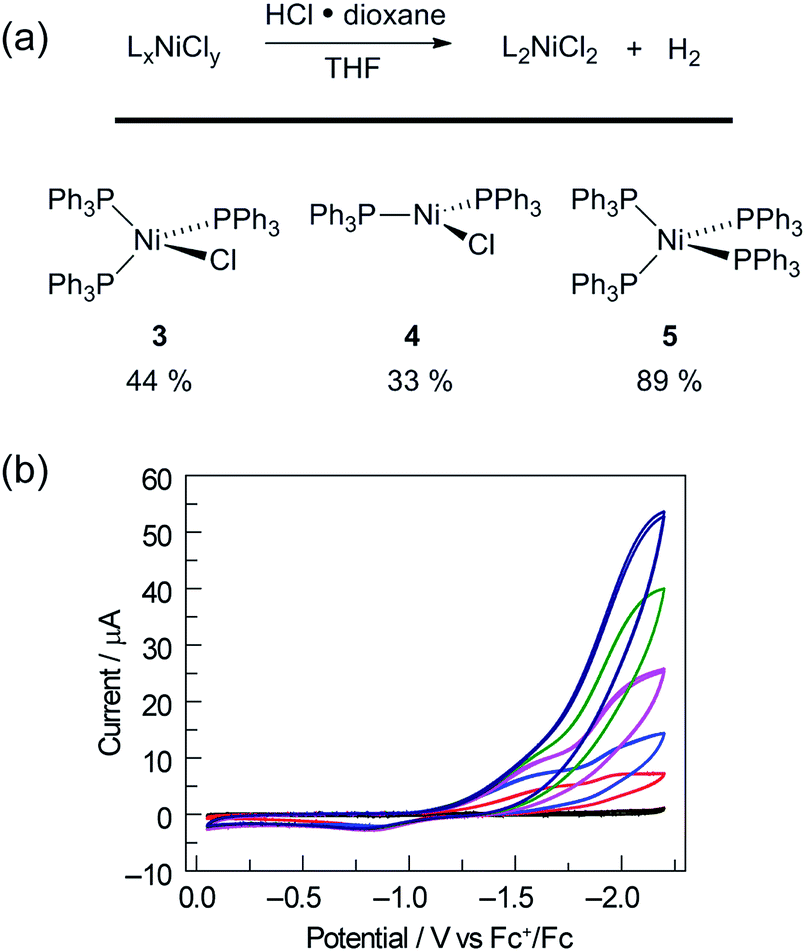 | ||
Fig. 4 (a) Protonation of Ni(I) and Ni(0) complexes afforded Ni(II)chloride as well as H2. (b) Electrochemical response of electrolyte background ( , black), 1 mM Ni complex 2[TEA] ( , black), 1 mM Ni complex 2[TEA] ( , red) to addition of HCl 1.0 equiv. ( , red) to addition of HCl 1.0 equiv. ( , blue), 5.0 equiv. ( , blue), 5.0 equiv. ( , pink), 9.0 equiv. ( , pink), 9.0 equiv. ( , green), 13.0 equiv. ( , green), 13.0 equiv. ( , dark blue) in CH3CN (0.1 M NBu4PF6; scan rate, 100 mV s−1). Glassy carbon working electrode, Ag/AgNO3 reference, and Pt wire counter electrode. , dark blue) in CH3CN (0.1 M NBu4PF6; scan rate, 100 mV s−1). Glassy carbon working electrode, Ag/AgNO3 reference, and Pt wire counter electrode. | ||
Conclusions
As is common for first-row transition metal complexes, nickel halide complexes typically exhibit very short excited state lifetimes. Direct photoactivation of M–X bonds using the molecular excited states of these complexes has proven challenging owing to their short lifetimes. To circumvent the limitations imposed by short excited state lifetimes, we have developed a tandem photoredox/transition metal catalysis approach to H2 evolution in which the chromophore and the H2-evolution catalyst are localized on different molecules. Using diaryl phosphines as photoredox mediators, we have demonstrated that relatively non-basic phosphines are capable of acting as photoredox mediators under acidic conditions. Robust photocatalytic systems have been developed by combining phosphine photoredox mediators and Ni phosphine H2-evolution catalysts. Time-resolved spectroscopy has revealed that phosphines serve as photochemical H-atom donors and activate the M–X bonds of Ni(II) halide complexes via halogen-atom abstraction. The H2-evolution catalytic cycle is closed by sequential disproportionation of Ni(I) to afford Ni(0) and Ni(II) and protolytic H2 evolution from the Ni(0) intermediate. The described photoredox strategy is attractive in that independent optimization of photoredox mediator and H2-evolution catalyst provides multiple handles for system optimization.Acknowledgements
We gratefully acknowledge Robert L. Halbach and D. Kwabena Bediako for helpful discussions, and the funding from NSF Grant CHE-1332783 and a Ruth L. Kirchenstein National Research Service award (F32GM103211) for D.C.P.Notes and references
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022 CrossRef CAS PubMed.
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001 CrossRef CAS PubMed.
- A. F. Heyduk and D. G. Nocera, Science, 2001, 293, 1639 CrossRef CAS PubMed.
- A. J. Esswein, A. S. Veige and D. G. Nocera, J. Am. Chem. Soc., 2005, 127, 16641 CrossRef CAS PubMed.
- N. Elgrishi, T. S. Teets, M. B. Chambers and D. G. Nocera, Chem. Commun., 2012, 48, 9474 RSC.
- T. R. Cook, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 28 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 7411 CrossRef CAS PubMed.
- T.-P. Lin and F. P. Gabbaï, J. Am. Chem. Soc., 2012, 134, 12230 CrossRef CAS PubMed.
- H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 10866 CrossRef CAS PubMed.
- W. E. Van Zyl, J. M. López-de-Luzuriaga, J. P. Fackler Jr and R. J. Staples, Can. J. Chem., 2001, 79, 896 CrossRef CAS.
- J. P. Fackler Jr, Inorg. Chem., 2002, 41, 6959 CrossRef PubMed.
- J. S. Ovens and D. B. Leznoff, Dalton Trans., 2011, 40, 4140 RSC.
- T. A. Perera, M. Masjedi and P. R. Sharp, Inorg. Chem., 2014, 53, 7608 CrossRef CAS PubMed.
- A. R. Karikachery, H. B. Lee, M. Masjedi, A. Ross, M. A. Moody, X. Cai, M. Chui, C. D. Hoff and P. R. Sharp, Inorg. Chem., 2013, 52, 4113 CrossRef PubMed.
- D. C. Powers, M. B. Chambers, T. S. Teets, N. Elgrishi, B. L. Anderson and D. G. Nocera, Chem. Sci., 2013, 4, 2880 RSC.
- E. I. Carrera, T. M. McCormick, M. J. Kapp, A. J. Lough and D. S. Seferos, Inorg. Chem., 2013, 52, 13779 CrossRef CAS PubMed.
- For a recent counter-example, see: D. C. Powers, S. J. Hwang, S.-L. Zheng and D. G. Nocera, Inorg. Chem., 2014, 53, 9122 CrossRef CAS PubMed.
- C. H. Lee, D. A. Lutterman and D. G. Nocera, Dalton Trans., 2013, 42, 2355 RSC.
- S. J. Tereniak, E. E. Marlier and C. C. Lu, Dalton Trans., 2012, 41, 7862 RSC.
- E. A. Juban, A. L. Smeigh, J. E. Monat and J. K. McCusker, Coord. Chem. Rev., 2010, 254, 2677 CrossRef PubMed.
- C. Creutz, M. Chou, T. L. Netzel, M. Okumura and N. Sutin, J. Am. Chem. Soc., 1980, 102, 1309 CrossRef CAS.
- C. H. Lee, T. R. Cook and D. G. Nocera, Inorg. Chem., 2011, 50, 714 CrossRef CAS PubMed.
- D. C. Powers, B. L. Anderson and D. G. Nocera, J. Am. Chem. Soc., 2013, 135, 18876 CrossRef CAS PubMed.
- K. Haav, J. Saame, A. Kütt and I. Leito, Eur. J. Org. Chem., 2012, 2167 CrossRef CAS.
- Y. Sakaguchi and H. Hayashi, J. Phys. Chem. A, 2004, 108, 3421 CrossRef CAS.
- D.-L. Versace, J. C. Bastida, C. Lorenzini, C. Cachet-Vivier, E. Renard, V. Langlois, J.-P. Malval, J.-P. Fouassier and J. Lalevée, Macromolecules, 2013, 46, 8808 CrossRef CAS.
- S. K. Wong, W. Sytnyk and J. K. S. Wan, Can. J. Chem., 1971, 49, 994 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- X.-F. Zhao and C. Zhang, Synthesis, 2007, 4, 551 Search PubMed.
- K. D. Schramm and J. A. Ibers, Inorg. Chem., 1980, 19, 2441 CrossRef CAS.
- T. Yano, M. Hoshino, M. Kuroboshi and H. Tanaka, Synlett, 2010, 5, 801 Search PubMed.
- M. C. Smith, S. C. Davies, D. L. Hughes and D. J. Evans, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, m509 CAS.
- A. Schmidpeter and S. Lochschmidt, Inorg. Synth., 1990, 27, 253 CrossRef CAS.
- G. D. Stucky, J. B. Folkers and T. J. Kistenmacher, Acta Crystallogr., 1967, 23, 1064 CrossRef CAS.
- D. D. Ellis and A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 1067 Search PubMed.
- N. C. Norman, A. G. Orpen, M. J. Quayle and G. R. Whittell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m160 Search PubMed.
- G. Bontempelli, F. Magno, M. D. Nobili and G. Schiavon, J. Chem. Soc., Dalton Trans., 1980, 2288 RSC.
- Y. Sakaguchi and H. Hayashi, Chem. Phys. Lett., 1995, 245, 591 CrossRef CAS.
- L. Maini, D. Braga, P. P. Mazzeo and B. Ventura, Dalton Trans., 2012, 41, 531 RSC.
- M. L. Kaufman and C. L. Griffin, Tetrahedron Lett., 1965, 12, 769 CrossRef.
- R. Waterman, Curr. Org. Chem., 2008, 12, 1322 CrossRef CAS.
- J. P. Roth, S. Lovell and J. M. Mayer, J. Am. Chem. Soc., 2000, 122, 5486 CrossRef CAS.
- L. J. J. Laarhoven, P. Mulder and D. D. M. Wayner, Acc. Chem. Res., 1999, 32, 342 CrossRef CAS.
- A. Cherkasov and M. Jonsson, J. Chem. Inf. Comput. Sci., 2000, 40, 1222 CrossRef CAS.
- W. van Scheppingen, E. Dorrestijn, I. Arends and P. Mulder, J. Phys. Chem. A, 1997, 101, 5404 CrossRef CAS.
- V. Fourmond, P.-A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and time-dependent photochemical data; transient absorption spectroscopy and electrochemical experimental details. CCDC 992218–992221. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02357a |
| ‡ Crystallographic data for 2[ClPPh3]: C36H30Cl4P2Ni, M = 725.05, orthorhombic, Pbca, a = 17.435(4), b = 15.662(3), c = 24.139(9), V = 6591(2), Z = 8, μ = 1.036 mm−1, T = 100(2) K, R1 = 0.0527, wR2 = 0.0720 (based on all reflections), GooF = 1.030, reflections measured = 57380, unique reflections = 5858, Rint = 0.0741. Crystallographic data for 6: C40H48Cl4O4P2Ni, M = 855.23, monoclinic, P21/n, a = 10.1261(8), b = 23.4579(18), c = 17.8192(14), β = 106.3550(12)°, V = 4061.4(5), Z = 4, μ = 0.859 mm−1, T = 100(2) K, R1 = 0.0788, wR2 = 0.1328 (based on all reflections), GooF = 1.005, reflections measured = 44051, unique reflections = 7212, Rint = 0.0668. Crystallographic data for Cl2PPh3: C18H15Cl2P, M = 333.17, monoclinic, P21/c, a = 13.338(3), b = 14.376(3), c = 8.7454(17), β = 102.53(3)°, V = 1637.0(6), Z = 4, μ = 0.484 mm−1, T = 100(2) K, R1 = 0.0330, wR2 = 0.0710 (based on all reflections), GooF = 1.060, reflections measured = 18333, unique reflections = 2874, Rint = 0.0308. Crystallographic data for [ClPPh3]OTf: C19H15ClF3O3PS, M = 446.79, monoclinic, P21/n, a = 11.255(2), b = 9.1501(18), c = 18.658(4), β = 93.04(3)°, V = 1918.7(7), Z = 4, μ = 0.438 mm−1, T = 100(2) K, R1 = 0.0425, wR2 = 0.0737 (based on all reflections), GooF = 1.031, reflections measured = 18058, unique reflections = 3392, Rint = 0.0362. |
| This journal is © The Royal Society of Chemistry 2015 |