
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pure-shift IMPRESS EXSIDE – Easy measurement of 1H–13C scalar coupling constants with increased sensitivity and resolution†
I. E.
Ndukwe
and
C. P.
Butts
*
School of Chemistry, University of Bristol, Cantocks Close, Bristol, BS8 1TS, UK. E-mail: craig.butts@bristol.ac.uk
First published on 7th December 2015
Abstract
The easy-to-interpret and rapid SelEXSIDE NMR experiment for measuring long-range 1H–13C scalar coupling constants nJCH is improved with increased sensitivity and resolution by the incorporation of ‘pure-shift’ homonuclear decoupling and IMPRESS-Hadamard encoding, with typical increases in signal-to-noise of over 300% and 400% demonstrated for small molecule examples, and narrower linewidths enabling measurement of smaller coupling constants.
NMR analyses of the 3D-structure of conformationally flexible small molecules in solution are often under-determined i.e. the stereochemistry or conformation cannot be definitively assigned because the number and accuracy of experimental parameters is too small to account for all of the possible configurations and conformations of the molecule. In organic molecules, 1H–1H scalar coupling constants (3JHH) and NOE measurements are the common NMR parameters used for this and are routinely combined with computational methods to improve structure elucidations or semi-quantitative methods exemplified by ‘J-based configurational analysis’ to acyclic systems.1–3 We have recently demonstrated that increased accuracy in quantitative NOE-distance analysis of small molecules4 allows finer details of dynamic 3D molecular structures to be resolved – for example identifying very small populations (∼2%) of molecular conformers,5 accurate determination of the rotational populations of a flexible chain6 and stereochemical elucidation of contiguous quaternary centres.7
Adding quantitative analysis of 2- and 3-bond 1H–13C scalar coupling constants (nJCH), of which there are many more than 3JHH couplings in most organic molecules, enables even greater levels of discrimination such that we have successfully described the strong helical or linear conformational preference of iteratively homologated alkane diastereomers, despite thousands of potential conformers.8 However, if chemists are to routinely use the potentially powerful nJCH couplings it is crucial that simple, reliable and accurate experimental and analysis methods must be available which are accessible to non-spectroscopists. The practical challenges of extracting nJCH values from NMR data include sensitivity, inability to measure values for quaternary carbons, and analysis of convoluted lineshapes.9,10 Most existing 2-dimensional methods rely on measuring 1H–13C coupling constants in the direct (horizontal) F2 dimension and suffer from simultaneous overlap of 1H–1H and 1H–13C couplings with severe lineshape distortions arising from this. Castanar et al. recently reported the ‘pure in-phase’ (PIP) HSQMBC11 which ensures clean 1H–1H and 1H–13C coupling co-evolution resulting in substantially improved F2 lineshapes, although complex 1H–1H multiplets still make it challenging to accurately measure small coupling constants (<2 Hz) with this approach.
An alternative approach to dealing with lineshape distortion is to use J-resolved approaches, exemplified by EXSIDE.12 Here the desired heteronuclear coupling constant is isolated and resolved as clean in-phase doublets in the indirect (vertical) F1 dimension of the spectrum (Fig. 1a). The F1-splitting of the peaks is the multiple of nJCH and a user-selected J-scaling factor, N. This provides an easy accurate means to measure even small nJCH values, down to ∼1 Hz in most cases. Such F1-based approaches, however, are relatively slow and weak and it generally requires several hours of measurement for each proton, while intensities are also reduced by T2 relaxation in the extended J-scaled INEPT preparation period. The long experiment time arises because a large number of t1 increments are needed to resolve small couplings in F1. Our recently reported 13C band-selective EXSIDE13 (SelEXSIDE) method substantially reduces the experiment time to just a few minutes by measuring each nJCH separately with a much narrower 13C window, thus needing fewer t1 increments. However the short experiment time also reduces signal averaging and thus sensitivity, so in practice it requires >10 mmol samples to obtain reasonable signal intensities using a 500 MHz room temperature NMR probehead.
 | ||
| Fig. 1 Strychnine structure and H11b–C10 correlations from (left-to-right): H20b/H11b EXSIDE spectrum (∼7.5 hours) with H11b–C10 inset; SelEXSIDE spectrum (50 minutes); ImpEXSIDE spectrum (50 minutes, as one of four correlations measured simultaneously); ImpEXSIDE_PS spectrum (50 minutes, as one of four correlations measured simultaneously). | ||
Herein we combine two methods to substantially improve the sensitivity of EXSIDE-based experiments: greater signal-averaging by spectrum aliasing and F1-Hadamard encoding; and increasing signal intensity (height) through F2-homonuclear decoupling.
Hadamard encoding of the F1 dimension of EXSIDE in 2D NMR spectra allows simultaneous measurement of multiple 13C regions of the EXSIDE spectrum – effectively simultaneous measurement of several SelEXSIDE13 experiments, providing greater signal averaging for each peak. Hadamard encoding is achieved by the IMPRESS technique described by Krishnamurthy,14 replacing SelEXSIDEs selective 13C inversion pulses with a series of n symmetrically shifted laminar pulses15 which are phase-encoded to match Hadamard matrices, Hn.14,16–18 The IMPRESS EXSIDE (ImpEXSIDE) pulse sequence (Fig. S1†)19 incorporates the 13C symmetrically shifted pulses (S1 and S2) during the DPFGSE refocussing period. S1 and S2 combine to phase encode the 13C chemical shift regions of interest. For a 4-region IMPRESS EXSIDE, in all scans S1 will always have an x phase at all four chemical shifts (Sxxxx), however the phase of S2 at each chemical shifts (A, B, C, D) will correspond to the rows of the H4 matrix shown below where ‘+’ and ‘−’ represent x and y phase pulses respectively at each chemical shift (that is S2 will be Sxxxx, Sxyxy, Sxxyy and Sxyyx for scans 1, 2, 3 and 4 respectively).

The desired sub-spectra for each 13C chemical shift region can then be readily extracted by the appropriate sum/difference of the four scans following the appropriate columns of the Hadamard matrix. For example, the sum of all four scans (Scan 1 + Scan 2 + Scan 3 + Scan 4) will give sub-spectrum A (while cancelling contributions from B, C and D) while Scan 1 + Scan 2 − Scan 3 − Scan 4 will give sub-spectrum C, etc (See ESI† for full details, along with pulse sequence and processing templates/macros for Varian/Agilent NMR spectrometers). The IMPRESS data for H11b–C10 of strychnine is shown in Fig. 1c alongside data for EXSIDE12 and SelEXSIDE13 experiments (Fig. 1a and b respectively).
ImpEXSIDE and SelEXSIDE data were obtained for all 13C couplings to H15a/b, H11b and H20b of strychnine. For comparability both techniques measured four nJCH values in 50 minutes (one 50 minute experiment for ImpEXSIDE, four 12.5 minute experiments for SelEXSIDE) and in all cases the nJCH values measured from these experiments matched to within 0.2 Hz, and were in line with literature experimental and computed values.13Fig. 2 illustrates the systematic increase in signal-to-noise ratio obtained with ImpEXSIDE (green) compared to the parent SelEXSIDE (blue). In fact in a number of cases correlations too weak to observe by SelEXSIDE under these conditions were clear in ImpEXSIDE experiments (H15a–C14, H15a–C16, H15b–C16, H15b–C12, H15b–C21, H20b–C18 and H20b–C22). In theory, the IMPRESS method should increase sensitivity for all correlations by around 2-fold compared to SelEXSIDE as signals are averaged for four-times longer but in practice a mean 2.5-fold increase in sensitivity was found, with some variation of responses from ∼1.5-fold (H20b–C21) to ∼4.0-fold (H11b–C12 and H15b–C13). This variability is ascribed to the differing placement of the selective 13C inversion pulses compared to SelEXSIDE (these are in the initial DPFGSE INEPT period for SelEXSIDE) and differential sum/difference decoding of the acquired FIDs, for example the all-plus column of a Hadamard matrix is more sensitive to instrumental errors.18 In any case, there is no doubt that substantial sensitivity improvements arise from the ImpEXSIDE as compared to the original SelEXSIDE when more than one nJCH value is required to be measured. Fig. 1 also highlights a reduction in the F1 half-height line-width moving from EXSIDE (20.2 Hz) to the faster selective variants ImpEXSIDE (13.4 Hz) and SelEXSIDE (11.0 Hz), allowing even better discrimination of smaller J-scaled couplings.
 | ||
| Fig. 2 The signal to noise ratio of peaks arising from heteronuclear coupling of H15a/b, H11b and H20b of strychnine from SelEXSIDE (blue), ImpEXSIDE (green) and Pure-shift ImpEXSIDE (red). | ||
Two practical limitations of ImpEXSIDE relate to the 1H and 13C selection frequencies. As with all EXSIDE techniques, the selected 1H region must not contain two protons that are mutually scalar coupled to ensure that only heteronuclear coupling is J-scaled in F1 (although multiple uncoupled protons can be selected if desired). Further, the selected 13C regions (A, B, C and D) must not overlap to any great extent as the phase-encoded pulse profiles for each region are not rectangular and imperfect phase behaviour will be observed in overlap regions – although there is no reason why multiple carbon peaks cannot be present in each selected 13C region.
Further improvement to the sensitivity of EXSIDE-based experiments can be made by eliminating 1H–1H coupling in F2 through gated band-selective refocussing of the homonuclear coupling during t2 acquisition (known variously as “pure-shift”, “HOBS” (HOmonuclear Band Selective) or “bash” (BAnd-Selective Homonuclear) decoupling methods).20–22 Broadly classified as ‘pure-shift’ spectra, these methods have raised a lot of interest recently23 with significant improvements in sensitivity where the 1H of interest (‘active’) is decoupled by selective inversion of their coupled partner (‘passive’) spins, by either band-selection22 or isotopic selection.24
Fig. 3 illustrates the band-selective J-refocussing element which is incorporated into the standard acquisition period of the ImpEXSIDE.19 The J-refocusing element (consisting of a hard 180° 1H pulse on all spins followed by a selective 180° 1H pulse on the active spin only) refocuses the t2 evolution of chemical shift, heteronuclear scalar coupling as well as homonuclear scalar coupling of the selected (active) 1H. Acquisition is systematically interrupted with this J-refocusing element after each acquired chunk of data. The length of each chunk, at/n (where at is acquisition time and n is number of repeats) should be <1/(3*nJHH),22 to minimize the breakthrough of homonuclear coupling. In practice, the choice of chunk length is largely determined by a compromise between resolution achieved and spectrum artifacts and in some cases can be as small as 1/(5*nJHH). When applied to ImpEXSIDE, the resulting correlations (Fig. 1d) are thus collapsed to simple singlets in F2. This decoupling method is also readily implemented in the parent EXSIDE and SelEXSIDE sequences with essentially the same improvements and limitations (see ESI† for details).
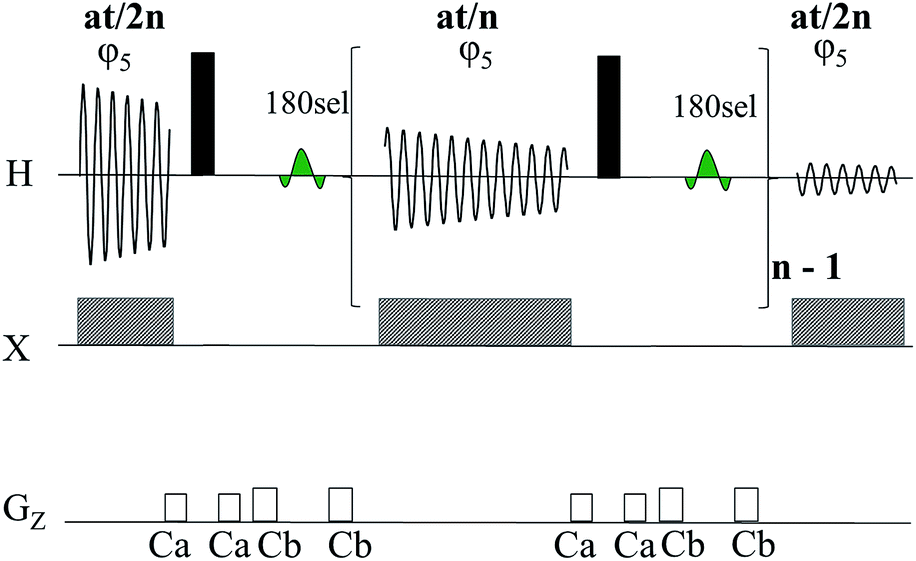 | ||
| Fig. 3 Band-selective J-refocussing element incorporated into the t2 acquisition period for band-selective homodecoupling. The 180sel pulse (green) is a refocussing 180° RSNOB. | ||
Importantly, this band-selective homonuclear decoupling can be incorporated without substantial loss of information or sensitivity as the EXSIDE-based experiments are already inherently F2(1H)-selective. Hence collapsing the F2-multiplets to narrow singlets should result in a substantial improvement in signal:noise (ideally up to 2-fold for a doublet or triplet, 4-fold for a resolved doublet of doublets, etc). On the other hand the F2-width of the final decoupled ‘singlet’ is limited to ∼4–6 Hz by B1 inhomogeneity of the pulses and the artificially increased T2 relaxation in the FID (resulting from non-acquisition during the J-refocusing period which, in some cases, contains relatively long 1H selective inversion pulses). Small F2-sideband artifacts appear at regular intervals equivalent to 1/(chunk length) either side of the decoupled resonance (visible in Fig. 1), arising from the regular discontinuities in the intensity of sequential data points where the FID is ‘stitched’ back together around the refocussing periods. Crucially, the presence of these low intensity F2 artefacts do not affect the measurement of the desired nJCH value in F1.
Fig. 2 illustrates the resulting 1.25–2.3 fold improvement in sensitivity of the Pure-shift modification (ImpEXSIDE_PS, red) on correlations for strychnine compared to the parent ImpEXSIDE (green). As expected, the largest increases were observed from the most highly coupled signal (H15a, doublet of triplets) while the smallest increases arise for the least coupled (H20b, doublet). It should be noted that strychnine is nearly a worst-case substrate for sensitivity improvement by homonuclear decoupling as its 1H spectrum consists mostly of sharp doublets/triplets. With more highly coupled substrates greater sensitivity enhancement can be achieved. For instance, application of ImpEXSIDE_PS to menthol (see ESI†) gave over 2-fold sensitivity enhancement compared to ImpEXSIDE in every case for the highly coupled signals of this more complex 1H NMR spectrum.
In summary, the sensitivity of EXSIDE and SelEXSIDE experiments can be substantially improved by the incorporation of IMPRESS (F1-spectral aliasing and Hadamard encoding) and F2-homonuclear decoupling either independently or in combination. In addition to improved resolution in both dimensions, there is no loss of signal or correlations from incorporating these modifications and >300% sensitivity increases are achieved for most resonances of strychnine, and substantially higher for more complex multiplets. With this sensitivity improvement, data acquisition should be an order of magnitude faster for sample-limited experiments and nJCH values should be measurable for single-digit mmol concentrations (corresponding to ∼1 mg of a 500 g mol−1 compound) in accessibly short experiment times.
Notes and references
- N. Matsumori, D. Kaneno, M. Murata, H. Nakamura and K. Tachibana, J. Org. Chem., 1999, 64, 866 CrossRef CAS PubMed.
- A. Ardá, M. I. Nieto, M. Blanco, C. Jiménez and J. Rodríguez, J. Org. Chem., 2010, 75, 7227 CrossRef PubMed.
- P. Cimino, G. Bifulco, A. Evidente, M. Abouzeid, E. Riccio and L. Gomez-Paloma, Org. Lett., 2002, 4, 2779 CrossRef CAS PubMed.
- C. P. Butts, C. R. Jones, E. C. Towers, J. L. Flynn, L. Appleby and N. J. Barron, Org. Biomol. Chem., 2011, 9, 177 CAS.
- C. P. Butts, C. R. Jones and J. N. Harvey, Chem. Commun., 2011, 47, 1193 RSC.
- C. R. Jones, C. P. Butts and J. N. Harvey, Beilstein J. Org. Chem., 2011, 7, 145 CrossRef CAS PubMed.
- M. G. Chini, C. R. Jones, A. D. Zampella, M. V. D'Auria, B. Renga, S. Fiorucci, C. P. Butts and G. Bifulco, J. Org. Chem., 2011, 77, 1489 CrossRef PubMed.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183 CrossRef CAS PubMed.
- B. L. Marquez, W. H. Gerwick and R. T. Williamson, Magn. Reson. Chem., 2001, 39, 499 CrossRef CAS.
- T. Parella and J. Felix Espinosa, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 17 CrossRef CAS PubMed.
- L. Castanar, J. Sauri, R. T. Williamson, A. Virgili and T. Parella, Angew. Chem., Int. Ed., 2014, 53, 8379 CrossRef CAS PubMed.
- V. V. Krishnamurthy, J. Magn. Reson., Ser. A, 1996, 121, 33 CrossRef CAS.
- C. P. Butts, B. Heise and G. Tatolo, Org. Lett., 2012, 14, 3256 CrossRef CAS PubMed.
- K. Krishnamurthy, J. Magn. Reson., 2001, 153, 124 CrossRef CAS PubMed.
- S. L. Patt, J. Magn. Reson., 1992, 96, 94 CAS.
- R. Crouch, R. D. Boyer, R. Johnson and K. Krishnamurthy, Magn. Reson. Chem., 2004, 42, 301 CrossRef CAS PubMed.
- K. Krishnamurthy, J. Magn. Reson., 2001, 153, 144 CrossRef CAS PubMed.
- E. Kupče, T. Nishida and R. Freeman, Prog. Nucl. Magn. Reson. Spectrosc., 2003, 42, 95 CrossRef.
- The pulse sequence diagram and a detailed walk-through can be found in the ESI†.
- R. W. Adams, L. Byrne, P. Kiraly, M. Foroozandeh, L. Paudel, M. Nilsson, J. Clayden and G. A. Morris, Chem. Commun., 2014, 50, 2512 RSC.
- L. Castañar, J. Saurí, P. Nolis, A. Virgili and T. Parella, J. Magn. Reson., 2014, 238, 63 CrossRef PubMed.
- N. H. Meyer and K. Zangger, Angew. Chem., Int. Ed., 2013, 52, 7143 CrossRef CAS PubMed.
- (a) K. Zangger, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 86–87, 1 CrossRef CAS PubMed; (b) L. Castanar, T. Parella Magn. Reson. Chem., 2015, 53, 399 CrossRef CAS PubMed.
- L. Paudel, R. W. Adams, P. Király, J. A. Aguilar, M. Foroozandeh, M. J. Cliff, M. Nilsson, P. Sándor, J. P. Waltho and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 11616 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, including pulse sequence diagrams, additional data acquired for menthol, and alternative acquisition modes for IMPRESS data and a tar file containing VNMRJ 4 compatible pulse sequence and acquisition/processing macros. See DOI: 10.1039/c5ra24926k |
| This journal is © The Royal Society of Chemistry 2015 |