
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot regioselective synthesis of functionalized and fused furans from Morita–Baylis–Hillman and Rauhut–Currier adducts of nitroalkenes†
Vaijinath Mane,
Tarun Kumar,
Sourav Pradhan,
Savita Katiyar and
Irishi N. N. Namboothiri*
Department of Chemistry, Indian Institute of Technology Bombay, Mumbai 400076, India. E-mail: irishi@iitb.ac.in; Fax: +91-22-2576-7152
First published on 10th August 2015
Abstract
Highly functionalized and fused furans have been synthesized via cascade reactions of Morita–Baylis–Hillman and Rauhut–Currier adducts of nitroalkenes with active methylene compounds. The reactions involving SN2′-intramolecular Michael addition or Michael addition-intra-molecular nucleophilic substitution take place in a regioselective manner to afford synthetically and biologically useful furans in moderate to good yields.
Introduction
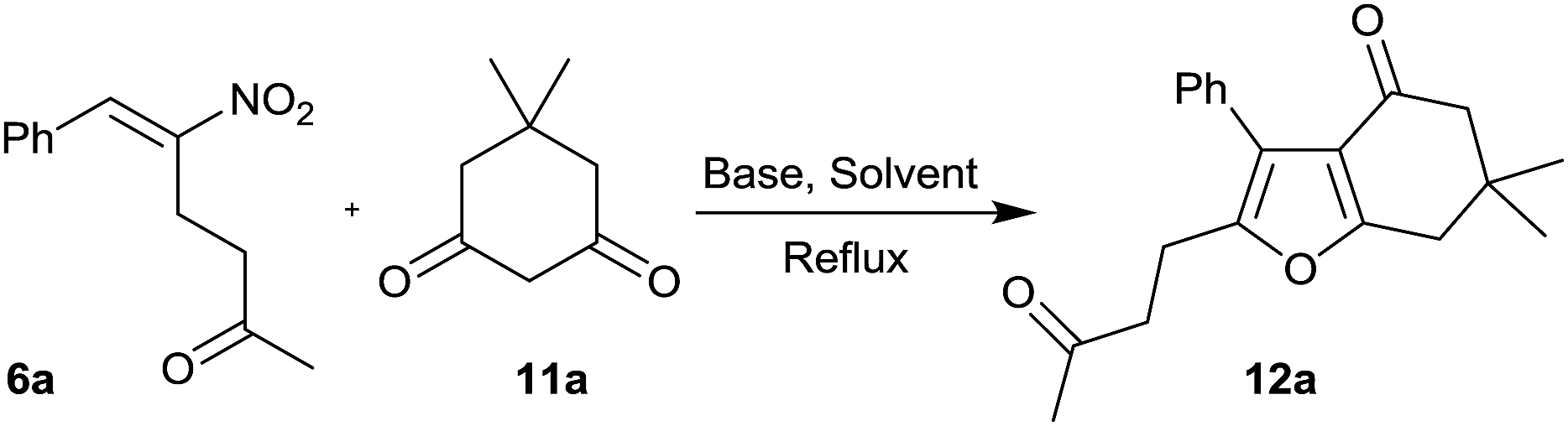
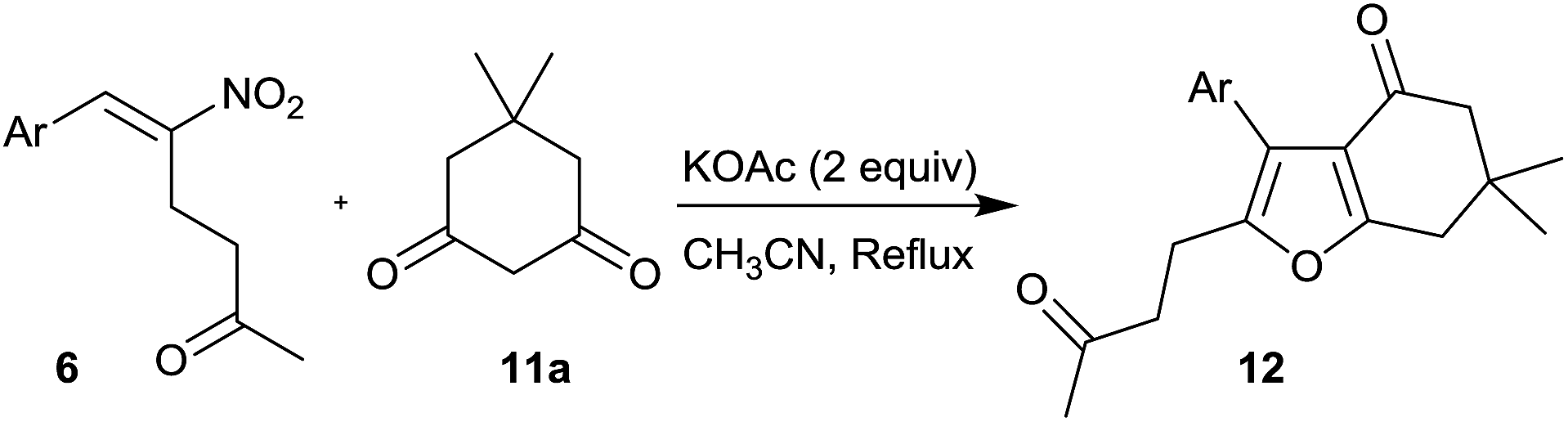
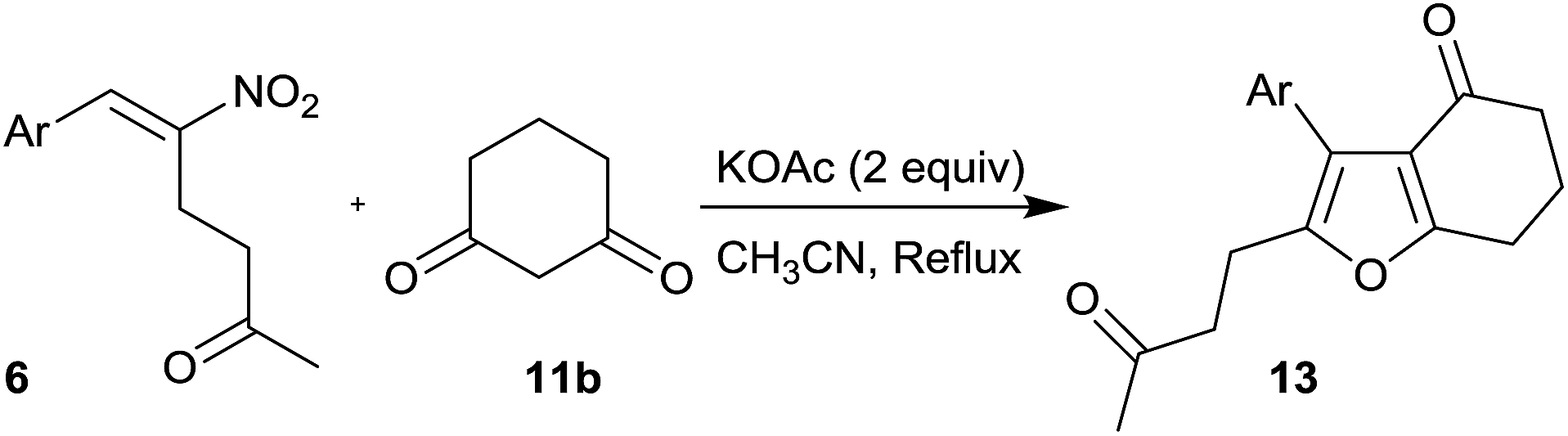
Furans belong to a unique class of five membered aromatic oxygen heterocycles which constitute the core structure of numerous natural products, drugs and other bioactive molecules.1 The diverse biological properties exhibited by furan containing compounds are well-documented in the literature.2 The wide applicability of furans as valuable synthons in multi-step reactions, including total synthesis, is well-recognized.3 Among the several methods reported for the synthesis of furans,4 Paal–Knorr synthesis (from 1,4-dicarbonyl compounds)5 and Feist–Benary synthesis (typically from α-haloketones and β-dicarbonyl compounds)6 are the prominent ones. Many highly efficient transition metal catalyzed cycloisomerization strategies have emerged in recent years.7Despite the availability of numerous methods, development of novel and efficient diversity oriented approaches for the synthesis of functionalized and fused furans would be very valuable both from synthetic and biological perspectives. As part of the studies on the Morita–Baylis–Hillman (MBH)8 and Rauhut–Currier (RC)9 reactions of nitroalkenes and the applications of the products,10–20 we and others have utilized the MBH acetates of nitroalkenes 1 (LG = OAc) for the synthesis of several carbocycles11 and heterocycles.12,13 The methodology involves a cascade SN2′ reaction of a binucleophile with MBH acetate 1 followed by an intramolecular Michael addition taking advantage of the 1,2 or 1,3-bi-electrophilic character of 1 as outlined in Scheme 1a. We and Chen et al. have reported the synthesis of furans via base mediated addition of 1,3-dicarbonyl compounds and arenols to the 1,2-bielectrophilic MBH acetates 1 (Scheme 1b).13 Herein we report the role of α-nitroacetophenone 4 as the bi-nucleophile towards the MBH acetates 1 resulting in synthetically and biologically useful highly substituted nitrofurans 5 as single regioisomers (Scheme 1c). The vinylogous MBH (Rauhut–Currier, RC) reaction of nitroalkenes14 and its applications for the synthesis of novel carbocyclic and heterocyclic scaffolds have also been of interest to us. We and others have employed RC adducts of nitroalkenes for the synthesis of functionalized pyrazoles,15 decalins,16 cycloalkanones,17 spirocycles18 and bridged heterobicyclics such as epibatidine.19 The reactivity profile of a representative RC adduct 6, derived from the imidazole/LiCl mediated RC reaction of nitroalkene with MVK, is outlined in Scheme 2a. Recently, we have reported a highly diastereoselective Michael initiated intramolecular aldol reaction involving compound 6 and cyclohexanone 7 for the synthesis of highly substituted trans-decalins 8 (Scheme 2b).16 The compound 6 reacts as a 1,4-electrophile-nucleophile in this case. Chen et al. have reported the formation of spirocyclic compound 10 upon addition of indane-1,3-dione 9 to compound 6 in which compound 6 reacted as a 1,5-bielectrophile.18 Surprisingly, when a 1,3-cyclohexanedione such as 11 (R = Me, dimedone) is added to compound 6, a new reactivity profile emerges in which compound 6 reacts as a 1,2-bielectrophile without the participation of the ketoalkyl moiety to provide fused furans 12, the results of which are also reported here.
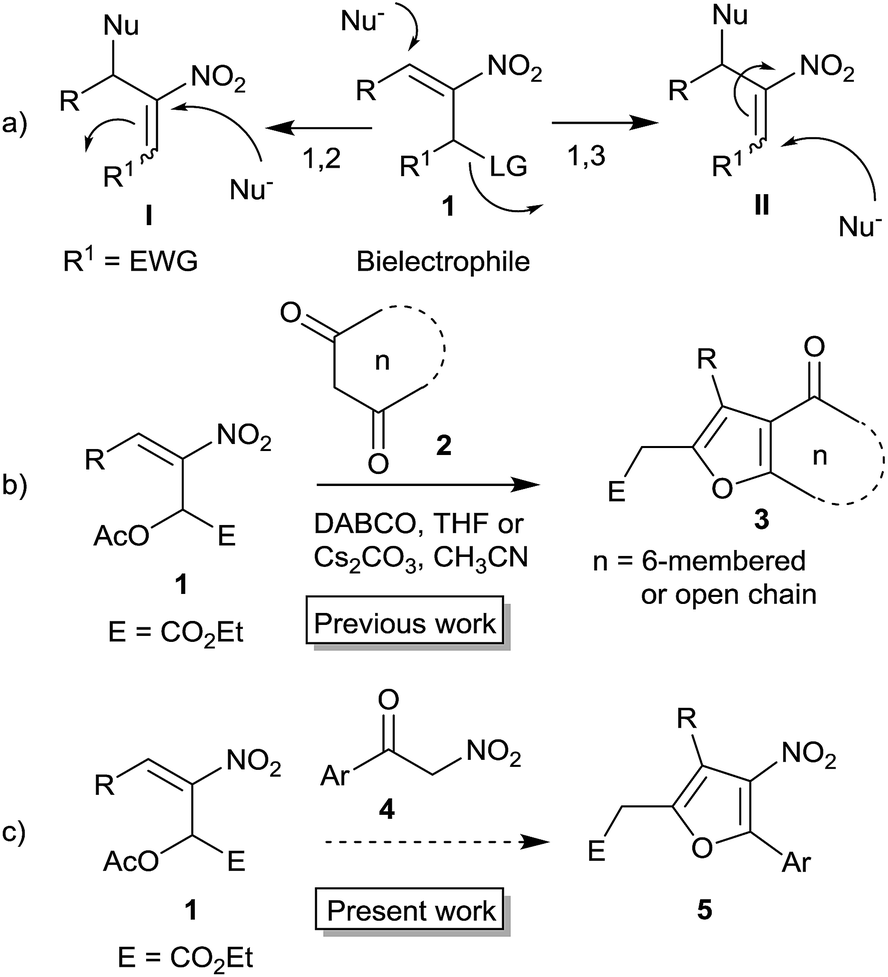 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
Results and discussion
Initially, acetate 1a and nitroketone 4 were selected as model substrates for our optimization studies for the synthesis of nitrofurans 5a (Table 1). In the presence of 1 equiv. of DABCO, at room temperature, in dichloromethane as solvent, we isolated furan 5a in low yield (17%, entry 1). The increase in the amount of base to 2 equiv. rendered the product 5a in improved yield (36%) and in lower reaction time (18 h, entry 2). Further improvement in the yield to 45% and decrease in reaction time to 12 h were observed when the solvent was changed to THF (entry 3). The reaction in the presence of a non-polar solvent such as toluene also resulted in low product yield (24%, entry 4). Next, we screened Et3N in THF which afforded the product only in low yield (22%, entry 5). However, the yield improved to 40% when the solvent was changed to dichloromethane (entry 6). In the same solvent (dichloromethane), various other organic amine bases such as diisopropylamine, Hünig's base, DMAP and DBU either provided lower yields or complex reaction mixtures (entries 7–10). The reaction in the presence of inorganic bases such as K2CO3 and Cs2CO3 also resulted in complex reaction mixtures (entries 11–13). Finally, the effect of temperature in the DABCO mediated reaction was investigated by carrying out the reaction in THF at elevated temperatures (45 °C and 65 °C, entries 14–15). Although the reaction rate improved, the yield remained unaffected at 45 °C (entries 3 and 14). At 65 °C, appreciable drop in the yield to 25% with considerable decomposition was observed (entry 15). Interestingly, the weakest base among the above, DABCO (pKa 8.7), was best suited for nitroacetophenone 4 (pKa 5.4), affording the product 5a in highest yield suggesting that slow generation of the enolate was desirable in our reaction.
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | % yieldb |
| a The reactions were carried out with 0.3 mmol each of 1a and 4 in 4 mL solvent.b After silica gel column chromatography.c 1 equivalent of DABCO was used.d Complex mixture.e Reaction temperature 45 °C.f Reaction temperature 65 °C. | ||||
| 1 | DABCOc | DCM | 26 | 17 |
| 2 | DABCO | DCM | 18 | 36 |
| 3 | DABCO | THF | 12 | 45 |
| 4 | DABCO | PhCH3 | 22 | 24 |
| 5 | NEt3 | THF | 20 | 22 |
| 6 | NEt3 | DCM | 22 | 40 |
| 7 | iPr2NH | DCM | 18 | 16 |
| 8 | iPr2EtN | DCM | 25 | 18 |
| 9 | DMAP | DCM | 24 | —d |
| 10 | DBU | DCM | 20 | 26 |
| 11 | Cs2CO3 | DCM | 22 | —d |
| 12 | Cs2CO3 | THF | 24 | —d |
| 13 | K2CO3 | DCM | 28 | —d |
| 14e | DABCO | THF | 9 | 44 |
| 15f | DABCO | THF | 6 | 25 |
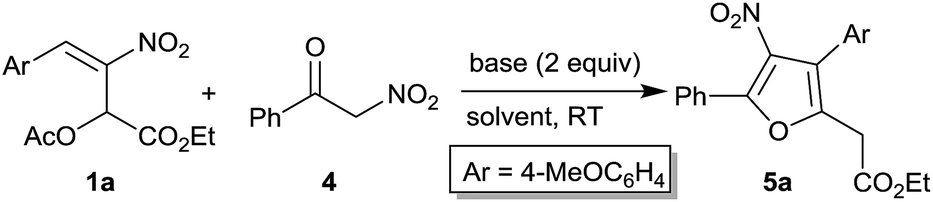
After screening different conditions, we identified DABCO as the best base and THF as the best solvent to afford the desired product 5a. Thus, the above optimized conditions, viz. 2 equiv. of DABCO, in THF, at room temperature (Table 1, entry 3), were employed to explore the scope of the reaction between different MBH acetates 1a–i and α-nitroacetophenone 4 (Table 2). Besides compound 1a which provided the product 5a in 45% yield (entry 1), MBH acetates bearing sterically and electronically diverse aryl groups 1b–i have been treated with compound 4 under our optimized conditions to afford tetrasubstituted furans 5b–i (entries 2–9). These include MBH acetate bearing parent phenyl group 1b, those bearing strongly electron donating substituents 1c,d and weakly electron donating substituent 1e, fused aromatic substituent 1f, weakly and strongly electron withdrawing substituents 1g,h and a heteroaryl substituent 1i. Although no appreciable substituent effect was discernible and the isolated yields of these nitrofurans 5 were consistently moderate (52–59%) in all the cases, the fact that such furans with well-defined substitution pattern could be easily synthesized from readily available compound 1 and compound 5a made our approach very attractive.
|
||||
|---|---|---|---|---|
| Entry | 1, Ar | Time (h) | 5 | % yieldb |
| a The reactions were carried out with 0.3 mmol each of 1 and 4 in 4 mL THF.b After silica gel column chromatography. No side products were observed in these reactions. | ||||
| 1 | 1a, 4-OMeC6H4 | 12 | 5a | 45 |
| 2 | 1b, C6H5 | 13 | 5b | 53 |
| 3 | 1c, 2,4-(OMe)2C6H3 | 7 | 5c | 57 |
| 4 | 1d, 3,4-(OCH2O)C6H3 | 8 | 5d | 57 |
| 5 | 1e, 4-MeC6H4 | 8 | 5e | 56 |
| 6 | 1f, 1-naphthyl | 7 | 5f | 59 |
| 7 | 1g, 4-ClC6H4 | 8 | 5g | 54 |
| 8 | 1h, 2-NO2C6H4 | 7 | 5h | 58 |
| 9 | 1i, 2-furyl | 9 | 5i | 52 |
The structure and regiochemistry of products 5a–i were confirmed by detailed spectral analysis. A peak for ester group at 1736–1740 cm−1 and two peaks for the nitro group at 1504–1519 and 1350–1366 were characteristic in the IR spectra. The protons of CH2 group attached to furyl appeared, in general, as singlets at δ 3.51–3.92 in 1H NMR. However, these protons appeared as AB spin systems in 5f and 5h which was attributable to atropisomerism due to restricted rotation about the C–C bond connecting the furyl moiety and the aryl group at position 3. Surprisingly, such a pattern is not observed in the case of 5c. The methylene carbon attached to furyl appeared at δ 32.8–34.0 in 13C NMR. Finally, the regiochemistry was unambiguously established from a medium NOE interaction observed between the deshielded anisyl protons (meta to methoxy) and the methylene protons attached furyl in 5a by 1H–1H NOESY experiment.
The proposed mechanism for the formation of highly substituted furans 5 is outlined in Scheme 3. DABCO mediated nucleophilic addition of α-nitroketone 4 to acetate 1 followed by elimination of acetate group in an overall SN2′ fashion generates intermediate III. A second enolization of the nitroketone moiety in III, facilitated by DABCO, followed by an intramolecular oxa-Michael addition in a 5-exo-trig fashion generates intermediate IV which further undergoes DABCO assisted elimination of HNO2 to afford highly substituted furans 5.
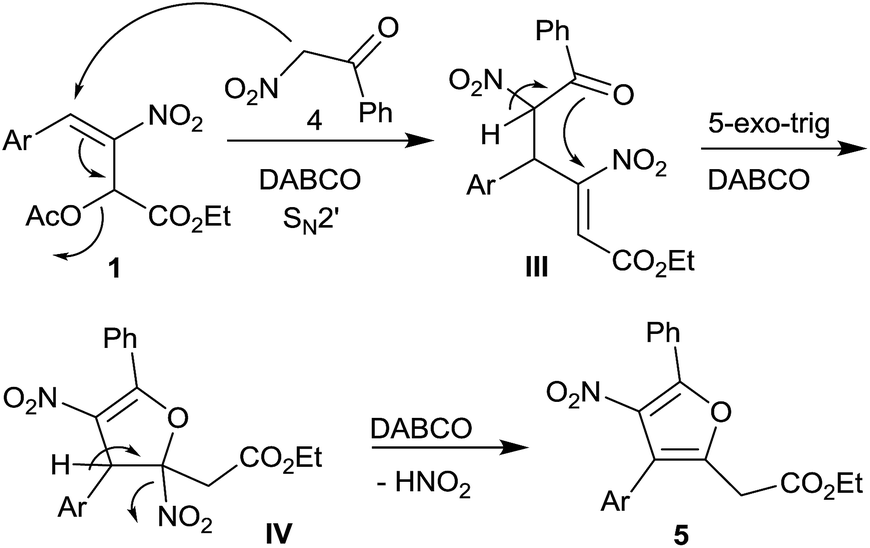 | ||
| Scheme 3 Plausible pathway for the formation of tetrasubstituted furans 5. | ||
Having synthesized a variety of nitrofurans 5 in satisfactory yield from the MBH acetates of nitroalkenes 1, we turned to RC adducts 6 as the key precursors for the synthesis of fused furans 12 as mentioned before (see Scheme 2) using six-membered cyclic 1,3-dicarbonyl compounds based on our own previous experience that 5-membered and 6-membered 1,3-dicarbonyls behaved differently in such reactions (see Scheme 1b).20
We have chosen compound 6a and compound 11a as the model substrates for our optimization studies (Table 3). When this reaction was performed in the presence of 1 equiv. of K2CO3 in CH3CN under reflux conditions, fused furan 12a was isolated in 23% yield within 9 h (entry 1). In order to improve the yield further, we screened various bases and solvents as summarized below.
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Solvent | Time (h) | % yieldb |
| a The reactions were carried out with 0.92 mmol of 6a and 0.92 mmol (1 equiv.) of 11a in 3 mL of solvent under reflux.b After silica gel column chromatography.c +20 mol% p-anisic acid.d +10 mol% TFA.e With acid work up.f Without acid work up. | ||||
| 1 | K2CO3 (1) | MeCN | 9 | 23 |
| 2 | DABCO (1) | MeCN | 10 | 15 |
| 3 | Et3N (1) | MeCN | 9 | 22 |
| 4 | Et3N (1)c | MeCN | 9 | 45 |
| 5 | DBU (1)d | MeCN | 3.5 | 20 |
| 6 | NaOAc (1) | MeCN | 9 | 29 |
| 7 | KOAc (1) | MeCN | 7 | 33 |
| 8 | KOAc (1)c | MeCN | 3.5 | 28 |
| 9 | KOAc (2) | MeCN | 8 | 42e |
| 10 | KOAc (2) | MeCN | 8 | 68f |
| 11 | KOAc (2) | THF | 8 | 42 |
| 12 | KOAc (2) | EtOH | 8 | 51 |
| 13 | KOAc (3) | MeCN | 7 | 35 |
The yields of furan 12a remained low (15–22%) when the reaction was conducted in CH3CN under reflux in the presence of 1 equiv. of different amine bases such as DABCO and Et3N (entries 2–3). Attempted improvement in the yield using Brønsted acid additives in conjunction with amine bases such as p-anisic acid and TFA met with only limited success (entries 4–5). Changing the base to NaOAc and KOAc, the latter even with p-anisic acid as additive, did not improve the yield (28–33%, entries 6–8). At this juncture, a reaction was carried out in the presence of 2 equiv. of KOAc which led to the formation of furan 12a in 42% yield (entry 9). This could be further improved to 68% by employing non-acidic workup conditions (entry 10). Other solvents such as THF and EtOH were less effective for our reaction (entries 11–12). Increasing the amount of base to 3 equiv. was detrimental as the yield dropped to 35% (entry 13).
As in the previous scheme (Tables 1 and 2), the weakest base, KOAc (pKa 4.7), appeared to give the best results in the addition of compound 11a (pKa 5.2) to compound 6a. The complex reaction pattern in the presence of stronger bases, including DABCO, leading to lower yields of the desired product 12a is attributable to side reactions involving the enolizable ketone side chain. Finally, the optimal conditions for the reaction, 2 equiv. of KOAc in CH3CN under reflux, were employed to study the scope of the reaction as reported in Tables 4 and 5.
|
||||
|---|---|---|---|---|
| Entry | 6, Ar | Time (h) | 12 | % yieldb |
| a The reactions were carried out with 0.92 mmol of 6, 0.92 mmol of 11a 1.80 mmol of KOAc in acetonitrile, reflux.b After silica-gel column chromatography. No side products were observed in these reactions. | ||||
| 1 | 6a, C6H5 | 9 | 12a | 68 |
| 2 | 6b, 4-MeC6H4 | 11 | 12b | 31 |
| 3 | 6c, 3-OMeC6H4 | 15 | 12c | 37 |
| 4 | 6d, 4-OMeC6H4 | 6 | 12d | 52 |
| 5 | 6e, 3,4-(OMe)2C6H3 | 4 | 12e | 37 |
| 6 | 6f, 3,4-(OCH2O)C6H3 | 5 | 12f | 34 |
| 7 | 6g, 4-ClC6H4 | 7 | 12g | 55 |
| 8 | 6h, 4-BrC6H4 | 2 | 12h | 35 |
| 9 | 6i, 2-thienyl | 15 | 12i | 29 |
| 10 | 6j, 1-naphthyl | 17 | 12j | 39 |
|
||||
|---|---|---|---|---|
| Entry | 6, Ar | Time (h) | 13 | % yieldb |
| a The reaction were carried out with 0.92 mmol of 6, 0.92 mmol of 11b and 1.80 mmol of KOAc in acetonitrile, reflux.b After silica-gel column chromatography; no side products were observed in these reactions. | ||||
| 1 | 6a, C6H5 | 12 | 13a | 33 |
| 2 | 6b, 4-MeC6H4 | 8 | 13b | 46 |
| 3 | 6c, 3-OMeC6H4 | 22 | 13c | 38 |
| 4 | 6d, 4-OMeC6H4 | 4 | 13d | 46 |
| 5 | 6e, 3,4-(OMe)2C6H3 | 4 | 13e | 30 |
| 6 | 6h, 4-BrC6H4 | 5 | 13f | 42 |
| 7 | 6i, 2-thienyl | 7 | 13g | 15 |
| 8 | 6j, 1-naphthyl | 20 | 13h | 44 |
The reaction of dimedone 11a with different RC adducts 6a–j was undertaken to obtain fused furans 12a–j (Table 4). Besides the RC adduct bearing parent phenyl group 6a which afforded the product 12a in 68% yield (entry 1), the RC adducts with aryl groups possessing a strongly electron donating substituent and a weakly electron withdrawing substituent at the para position, 6d and 6g, respectively, delivered the corresponding products 13d and 13g in decent yields (52% and 55%, entries 4 and 7). The yields of fused furans from other RC adducts possessing various electron donating and withdrawing substituents on the aromatic ring viz. 6b,c, 6e,f and 6h, remained low (31–37%, entries 2–3, 5–6 and 8). While low yield of the product 12i was encountered in the case of heteroaryl compound 6i (entry 9), the product yield was moderate (39%) in the case of a fused arylated compound 6j (entry 10).
After the successful demonstration of the reactivity of different types of RC adducts 6 with dimedone 11a, we focused our attention for the reaction of 1,3-cyclohexanedione 11b with different types of RC adducts 6 and the results are summarized in Table 5.
In this reaction, when Ar was Ph, the product was formed in poor yield (33%, entry 1) and electron donating groups on the phenyl ring of RC adducts provided poor to moderate yield of the products (30–46%, entry 2–5). RC adducts bearing bromo and naphthyl substituents also provided moderate yields (42–44%, entries 6 and 8) and in the case of heteroaryl substituent, a substantial decrease in the yield to 15% was observed (entry 7).
It may be noted that the reaction of 6i with 11a and 11b afforded the corresponding products 12i and 13g in lowest yields (29% and 15%, respectively, Table 4, entry 9 and Table 5 entry 7). This may be due to side reactions such as intermolecular Diels–Alder reaction between the thiophene moiety as the diene and the nitroalkene moiety as the dienophile under our experimental conditions.
As in the case of 5, the structure and regiochemistry of products 12 and 13 were established by analysis of their spectral data. While the saturated carbonyl appears in IR at 1713–1717 cm−1, the unsaturated carbonyl appears at 1672–1679 cm−1. The two isolated methylene protons in 12 appear, in general, as singlets at δ 2.34–2.36 and 2.71–2.74. The corresponding protons in 12j appear as AB spin system presumably due to atropisomerism about the C–C bond connecting the furyl and the aryl group at position 3. The regiochemistry was amply evident from a medium NOE interaction between the phenyl protons and the deshielded methylene protons of the oxobutyl side chain in 12a.
Scheme 4 outlines the plausible mechanism for the one-pot synthesis of furan by the present protocol. KOAc mediated Michael addition of 1,3-dicarbonyl compound 11 to compound 6 affords intermediate V. In the next step, an intramolecular oxa-Mannich type reaction takes place in a 5-exo-trig manner affording the intermediate VI, which on elimination affords the desired product 12 or 13.21
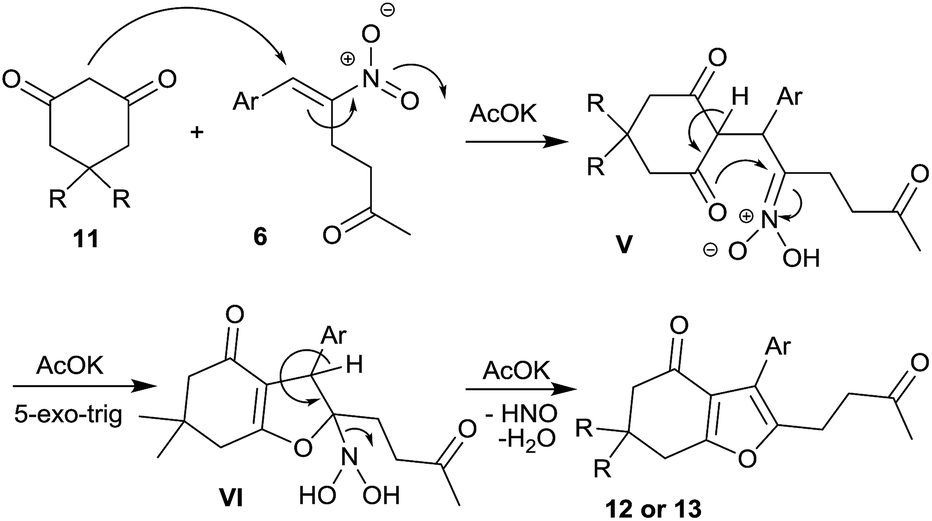 | ||
| Scheme 4 Plausible mechanism of furan formation. | ||
Conclusions
Novel methods for the synthesis of highly substituted furans from the Morita–Baylis–Hillman and Rauhut–Currier adducts of nitroalkenes have been developed. These include a cascade SN2′-intramolecular oxa-Michael addition and a cascade Michael-intramolecular oxa-Mannich reaction. Although the yields in these reactions are moderate, our approach is distinguished by the convenient access to highly functionalized and fused furans from readily available MBH and RC adducts of nitroalkenes.Experimental section
General experimental details
The melting points recorded are uncorrected. NMR spectra (1H, 1H decoupled 13C and 1H–1H NOESY) were recorded with TMS as the internal standard. The coupling constants (J values) are given in Hz. High resolution mass spectra were recorded under ESI Q-TOF conditions. MBH acetates 1,22 nitroacetophenone 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 and RC adducts 6 (ref. 14) were prepared by literature methods.
23 and RC adducts 6 (ref. 14) were prepared by literature methods.
General procedure for the preparation of tetrasubstituted furans 5 from MBH acetates 1 and α-nitroacetophenone 4
To a stirred solution of compound 1 (0.1 mmol) and compound 4 (33 mg, 0.2 mmol, 2 equiv.) in THF (3 mL) at room temperature was added DABCO (22 mg, 0.2 mmol, 2 equiv.) and the stirring was continued till the completion of the reaction (monitored by TLC, see Table 2). The reaction mixture was concentrated in vacuo and the crude residue was purified by silica gel column chromatography by eluting with 4–10% ethyl acetate/petroleum ether (gradient elution).Ethyl-2-(3-(4-methoxyphenyl)-4-nitro-5-phenylfuran-2-yl)acetate (5a)
Yellow oily liquid; yield 17 mg, 45%; IR (KBr, cm−1) 1739 (s), 1514 (vs.), 1360 (m); 1H NMR (400 MHz, CDCl3) δ 7.81–7.84 (m, 2H), 7.46–7.50 (m, 3H), 7.28 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.8 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 3.84 (s, 3H), 3.65 (s, 2H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.3, 32.8, 55.4, 61.8, 114.2, 121.2, 121.3, 127.6, 128.3, 128.6, 130.6, 130.9, 131.4, 144.8, 151.4, 159.8, 168.6; MS (ES+, Ar) m/z (rel intensity) 404 (MNa+, 30), 399 ([M + H2O]+,49), 382 (MH+, 100); HRMS (ES+, Ar) calcd for C21H20NO6 (MH+) 382.1291, found 382.1301. Confirmed by 1H–1H NOESY experiment.Ethyl-2-(4-nitro-3,5-diphenylfuran-2-yl)acetate (5b)
Yellow oily liquid; yield 19 mg, 53%; IR (KBr, cm−1) 1740 (vs.), 1513 (m), 1358 (m); 1H NMR (400 MHz, CDCl3) δ 7.81–7.84 (m, 2H), 7.46–7.50 (m, 3H), 7.41–7.45 (m, 3H), 7.34–7.37 (m, 3H), 4.20 (q, J = 7.1 Hz, 2H), 3.66 (s, 2H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.3, 32.9, 61.8, 121.8, 127.6, 128.4, 128.7, 128.7, 128.8, 129.3, 129.7, 130.7, 134.6, 144.9, 151.6, 168.5; MS (ES+) m/z (rel intensity) 413 ([M + Na + K]+, 100), 390 (MK+, 35), 374 (MNa+, 91), 343 (15); HRMS (ESI) calcd for C20H17NNaO5 (MNa+) 374.0999, found 374.1002.Ethyl-2-(3-(2,4-dimethoxyphenyl)-4-nitro-5-phenylfuran-2-yl)acetate (5c)
Yellow oily liquid; yield 23 mg, 57%; IR (KBr, cm−1) 1738 (vs.), 1511 (s), 1366 (m); 1H NMR (400 MHz, CDCl3) δ 7.83–7.80 (m, 2H), 7.48–7.45 (m, 3H), 7.24 (d, J = 8.3 Hz, 1H), 6.57 (dd, J = 8.3, 2.3 Hz, 1H), 6.52 (d, J = 2.3 Hz, 1H), 4.20 (q, J = 7.1 Hz, 2H), 3.85 (s, 3H), 3.72 (s, 3H), 3.65 (s, 2H), 1.29 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.3, 33.0, 55.5, 55.6, 61.8, 99.1, 104.8, 111.0, 118.1, 127.8, 128.4, 128.6, 130.4, 131.1, 144.5, 150.9, 158.3, 161.6, 168.8; MS (ES+) m/z (rel intensity) 450 (MK+, 69), 434 (MNa+, 78), 413 ([M + 2]+, 100), 391 (10); HRMS (ES+) calcd for C22H21NNaO7 (MNa+) 434.1210, found 434.1211.Ethyl-2-(3-(benzo[d][1,3]dioxol-5-yl)-4-nitro-5-phenylfuran-2-yl)acetate (5d)
Light yellow oily liquid; yield 67 mg, 57%; IR (KBr, cm−1) 1738 (vs.), 1504 (vs.), 1359 (s); 1H NMR (400 MHz, CDCl3) δ 7.81–7.79 (m, 2H), 7.48–7.46 (m, 3H), 7.25 (s, 1H), 6.88–6.82 (m, 3H), 6.02 (s, 1H), 4.23–4.18 (q, J = 7.2 Hz, 2H), 3.65 (s, 2H), 1.28 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.3, 32.8, 61.9, 101.5, 108.6, 110.2, 121.4, 122.6, 123.4, 127.6, 128.4, 128.7, 130.7, 145.0, 148.0, 148.1, 151.4, 168.5; MS (ES+) m/z (rel intensity) 434 (MK+, 60), 418 (MNa+, 100), 393 ([M − 2]+, 51), 355 (11), 331 (10), 307 (9); HRMS (ES+) calcd for C21H17NNaO7 (MNa+) 418.0897, found 418.0898.Ethyl-2-(4-nitro-5-phenyl-3-p-tolylfuran-2-yl)acetate (5e)
Light yellow oily liquid; yield 61 mg, 56%; IR (KBr, cm−1) 1738 (vs.), 1513, 1358 (m); 1H NMR (400 MHz, CDCl3) δ 7.83–7.80 (m, 2H), 7.44–7.41 (m, 3H), 7.29–7.21 (m, 4H), 4.20 (q, J = 7.2 Hz, 2H), 3.65 (s, 2H), 2.41 (s, 3H), 1.28 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.3, 21.5, 32.9, 61.8, 121.7, 126.2, 127.7, 128.4, 128.7, 129.3, 129.5, 130.4, 130.6, 138.6, 144.8, 151.5, 168.6; MS (ES+) m/z (rel intensity) 404 (MK+, 23), 388 (MNa+, 100), 366 (MH+, 25), 301 (20); HRMS (ES+) calcd for C21H19NNaO5 (MNa+) 388.1155, found 388.1155.Ethyl-2-(3-(naphthalen-1-yl)-4-nitro-5-phenylfuran-2-yl)acetate (5f)
Light yellow oily liquid; yield 71 mg (59%); IR (KBr, cm−1) 1739 (vs.), 1510 (vs.), 1354 (m); 1H NMR (400 MHz, CDCl3) δ 7.96–7.92 (m, 4H), 7.75 (d, J = 8.0 Hz, 1H), 7.57–7.46 (m, 7H), 4.14–4.08 (q, J = 7.2 Hz, 2H), 3.59, 3.53 (AB, J = 17.0 Hz, 2H), 1.19 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.2, 32.8, 61.7, 119.9, 125.1, 125.5, 126.4, 126.9, 127.7, 128.4, 128.7 (× 2), 128.7, 129.5, 130.8, 132.5, 133.7, 135.4, 145.9, 152.2, 168.3; MS (ES+) m/z (rel intensity) 440 (MK+, 77), 424 (MNa+, 100), 397 (20), 357 (23), 301 (35), 243 (69), 213 (100); HRMS (ES+) calcd for C24H19NNaO5 (MNa+) 424.1155, found 424.1154.Ethyl-2-(3-(4-chlorophenyl)-4-nitro-5-phenylfuran-2-yl)acetate (5g)
Light yellow oily liquid; yield 62 mg, 54%; IR (KBr, cm−1) 1739 (m), 1514 (m), 1358 (w), 739 (vs.); 1H NMR (400 MHz, CDCl3) δ 7.83–7.80 (m, 2H), 7.50–7.47 (m, 3H), 7.42, 7.31 (AB, J = 8.5 Hz, 4H), 4.20 (q, J = 7.1 Hz, 2H), 3.63 (s, 2H), 1.28 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.3, 32.8, 62.0, 120.8, 127.5, 127.8, 128.6, 128.7, 129.0, 130.9, 131.1, 134.9, 145.1, 152.1, 168.4; MS (ES+) m/z (rel intensity) 426 ([M + 2]K+, 10), 424 (MK+, 30), 413 (100), 410 ([M + 2]Na+, 5), 408 (MNa+, 15), 386 (MH+, 80); HRMS (ES+) calcd for C20H17ClNO5 (MH+) 386.0790, found 386.0784.Ethyl-2-(4-nitro-3-(2-nitrophenyl)-5-phenylfuran-2-yl)acetate (5h)
Light yellow oily liquid; yield 69 mg, 58%, mixture of atropisomers; IR (KBr, cm−1) 1736 (vs.), 1527 (vs.), 1512 (vs.), 1350 (vs.); 1H NMR (400 MHz, CDCl3) δ 8.26–8.22 (m, 1H), 8.14–8.10 (m, 0.5H, due to atropisomerism), 7.90–7.86 (m, 2H), 7.74–7.69 (m, 1H), 7.66–7.60 (m, 1.5H, due to atropisomerism), 7.53–7.46 (m, 4 + 1H), 4.15 (q, J = 7.1 Hz, 2H), 3.62, 3.51 (AB, J = 16.9 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.2, 33.0, 62.1, 118.8, 125.4, 127.4, 128.6, 129.2, 130.2, 130.4, 131.1, 132.9, 133.7, 134.4, 144.8, 148.8, 153.2, 168.0; MS (ES+) m/z (rel intensity) 435 (MK+, 16), 419 (MNa+, 100), 397 (14), 303 (23); HRMS (ES+) calcd for C20H16N2NaO7 (MNa+) 419.0850, found 419.0848.Ethyl-2-(4′-nitro-5′-phenyl-2,3′-bifuran-2′-yl)acetate (5i)
Yellow oily liquid; yield 18 mg (52%); IR (KBr, cm−1) 1739 (vs.), 1519 (s); 1H NMR (400 MHz, CDCl3) δ 7.78–7.74 (m, 2H), 7.50 (dd, J = 1.8, 0.6 Hz, 1H), 7.49–7.45 (m, 3H), 6.67 (dd, J = 3.4, 0.6 Hz, 1H), 6.50 (m, J = 3.4, 1.8 Hz, 1H), 4.20 (q, J = 7.1 Hz, 2H), 3.92 (s, 2H), 1.28 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 14.3, 34.0, 42.5, 61.9, 110.9, 111.5, 127.2, 128.3, 128.8, 130.8, 142.8, 143.2, 145.5, 151.8, 168.4; MS (ES+) m/z (rel intensity) 380 (MK+, 34), 364 (MNa+, 58), 360 (39), 349 (100); HRMS (ES+) calcd for C18H15NNaO6 (MNa+) 364.0792, found 364.0792.Representative procedure for the synthesis of substituted furans 12 and 13
To a stirred solution of 1,3-diketone 11 (0.92 mmol) in MeCN (3 mL) was added potassium acetate (0.180 g, 1.84 mmol, 2 equiv.) followed by Rauhut–Currier adduct 6 (0.92 mmol, 1 equiv.). The resulting reaction mixture was refluxed till the completion of the reaction. After the completion of the reaction (monitored by TLC, see Tables 4 and 5), the reaction mixture was concentrated in vacuo and the crude residue was purified by silica gel column chromatography (pet ether:ethyl acetate 5–10%, gradient elution).
6,6-Dimethyl-2-(3-oxobutyl)-3-phenyl-6,7-dihydrobenzofuran-4(5H)-one (12a)
White solid; yield 194 mg, 68%; mp 88–89 °C; IR (KBr, cm−1) 1717 (s), 1678 (vs.); 1H NMR (CDCl3, 400 MHz) δ 7.39–7.28 (m, 5H), 2.93 (t, J = 7.0 Hz, 2H), 2.78 (t, J = 7.0 Hz, 2H), 2.74 (s, 2H), 2.36 (s, 2H), 2.13 (s, 3H), 1.15 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 20.5, 28.7, 30.0, 35.1, 37.8, 41.8, 53.1, 118.7, 119.6, 127.4, 128.1, 129.9, 131.4, 151.2, 165.2, 193.6, 207.1; MS (ES+) m/z (rel intensity) 333 (MNa+, 75), 311 (MH+, 100), 254 (6), 253 (29); HRMS (ES+) calcd for C20H23O3 (MH+) 311.1647, found 311.1651.3-(4-Methylphenyl)-6,6-dimethyl-2-(3-oxobutyl)-6,7-dihydro-benzenefuran-4(5H)-one (12b)
Brown solid; yield 92 mg, 31%; mp 128–130 °C; IR (KBr, cm−1) 1717 (s), 1677 (vs.); 1H NMR (CDCl3, 400 MHz) δ 7.25, 7.18 (AB, J = 7.9 Hz, 4H), 2.93 (t, J = 7.1 Hz, 2H), 2.77 (t, J = 7.1 Hz, 2H), 2.73 (s, 2H), 2.36 (s, 3H), 2.35 (s, 2H), 2.13 (s, 3H), 1.15 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 20.5, 21.4, 28.8, 30.0, 35.1, 37.8, 41.8, 53.2, 118.8, 119.5, 128.3, 128.9, 129.8, 137.1, 151.0, 165.1, 193.6, 207.1; MS (ES+) m/z (rel intensity) 347 (MNa+, 38), 325 (MH+, 100), 268 (10), 267 (48); HRMS (ES+) calcd for C21H25O3 (MH+), 325.1804, found 325.1797.3-(3-Methoxyphenyl)-6,6-dimethyl-2-(3-oxobutyl)-6,7-dihydro-benzofuran-4(5H)-one (12c)
Yellow brown amorphous solid; yield 116 mg, 37%; IR (KBr, cm−1) 1714 (s), 1678 (vs.); 1H NMR (CDCl3, 500 MHz) δ 7.28 (t, J = 7.9 Hz, 1H), 6.94 (d, J = 2.2 Hz, 1H), 6.93 (d, J = 7.9 Hz, 1H), 6.85 (dd, J = 7.9, 2.4 Hz, 1H), 3.82 (s, 3H), 2.94 (t, J = 7.5 Hz, 2H), 2.77 (t, J = 7.5 Hz, 2H), 2.73 (s, 2H), 2.36 (s, 2H), 2.13 (s, 3H), 1.15 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 20.6, 28.8, 30.0, 35.1, 37.8, 41.8, 53.2, 55.4, 113.3, 115.7, 119.5, 122.4, 129.1, 132.7, 151.3, 159.3, 165.2, 193.5, 207.1; MS (ES+) m/z (rel intensity) 379 (MK+, 100), 363 (MNa+, 30), 341 (MH+, 5); HRMS (ES+) calcd for C21H24O4K (MK+), 379.1306, found 379.1306.3-(4-Methoxyphenyl)-6,6-dimethyl-2-(3-oxobutyl)-6,7-dihydrobenzofuran-4(5H)-one (12d)
Yellow solid; yield 163 mg, 52%; mp 109–110 °C; IR (KBr, cm−1) 1716 (s), 1675 (vs.); 1H NMR (CDCl3, 400 MHz) δ 7.29 (d, J = 9.5 Hz, 2H), 6.91 (d, J = 9.5 Hz, 2H), 3.82 (s, 3H), 2.92 (t, J = 7.1 Hz, 2H), 2.77 (t, J = 7.1 Hz, 2H), 2.72 (s, 2H), 2.35 (s, 2H), 2.13 (s, 3H), 1.15 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 20.5, 28.7, 30.0, 35.0, 37.8, 41.8, 53.1, 55.4, 113.6, 118.7, 119.2, 123.6, 131.0, 150.8, 159.0, 165.1, 193.7, 207.2; MS (ES+) m/z (rel intensity) 363 (MNa+, 60), 341 (MH+, 100), 284 (19), 283 (85), 190 (4); HRMS (ES+) calcd for C21H25O4 (MH+), 341.1753, found 341.1750.3-(3,4-Dimethoxyphenyl)-6,6-dimethyl-2-(3-oxobutyl)-6,7-dihydrobenzofuran-4(5H)-one (12e)
Yellow solid; yield 126 mg, 37%; 112–113 °C; IR (KBr, cm−1) 1716 (s), 1676 (s); 1H NMR (CDCl3, 400 MHz) δ 7.00 (s, 1H), 6.89, 6.86 (AB, J = 8.2 Hz, 2H), 3.89 (s, 3H), 3.88 (s, 3H), 2.94 (t, J = 7.8 Hz, 2H), 2.79 (t, J = 7.8 Hz, 2H), 2.72 (s, 2H), 2.35 (s, 2H), 2.13 (s, 3H), 1.14 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 20.5, 28.6, 29.9, 34.9, 37.7, 41.5, 53.1, 55.9, 56.0, 110.7, 113.6, 118.5, 119.2, 122.0, 123.8, 148.3, 150.7, 165.1, 193.6, 207.0; MS (ES+) m/z (rel intensity) 393 (MNa+, 10), 371 (MH+, 100), 314 (7), 313 (29); HRMS (ES+) calcd for C22H27O5 (MH+), 371.1858, found 371.1873.3-(Benzo[d][1,3]dioxol-5-yl)-6,6-dimethyl-2-(3-oxobutyl)-6,7-dihydrobenzofuran-4(5H)-one (12f)
Yellow solid; yield 111 mg, 34%; mp 109–111 °C; IR (KBr, cm−1) 1716 (s), 1677 (vs.); 1H (CDCl3, 400 MHz) δ 6.84 (s, 1H), 6.81–6.77 (m, unresolved AB, 2H), 5.95 (s, 2H), 2.90 (t, J = 7.4 Hz, 2H), 2.76 (t, J = 7.4 Hz, 2H), 2.71 (s, 2H), 2.34 (s, 2H), 2.13 (s, 3H), 1.14 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 20.5, 28.7, 30.0, 35.0, 37.8, 41.7, 53.1, 101.1, 108.1, 110.6, 118.7, 119.3, 123.3, 125.0, 147.0, 147.4, 151.0, 165.1, 193.6, 207.1; MS (ES+) m/z (rel intensity) 393 (MK+, 100), 377 (MNa+, 25); HRMS (ES+) calcd for C21H22O5K (MK+) 393.1099, found 393.1098.3-(4-Chlorophenyl)-6,6-dimethyl-2-(3-oxobutyl)-6,7-dihydro-benzofuran-4(5H)-one (12g)
White solid; yield 174 mg, 55%; mp 86–88 °C; IR (KBr, cm−1) 1713 (s), 1672 (vs.), 737 (s); 1H NMR (CDCl3, 400 MHz) δ 7.34, 7.31 (AB, J = 8.6 Hz, 4H), 2.90 (t, J = 7.4 Hz, 2H), 2.78 (t, J = 7.4 Hz, 2H), 2.73 (s, 2H), 2.35 (s, 2H), 2.14 (s, 3H), 1.15 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 20.4, 28.7, 30.0, 35.1, 37.8, 41.5, 53.1, 118.5, 118.6, 128.3, 129.9, 131.3, 133.4, 151.4, 165.4, 193.7, 206.9; MS (ES+) m/z (rel intensity) 347 ([M + 2]H+, 35), 345 (MH+, 100), 289 (27), 287 (81), 242 (4), 192 (7); HRMS (ES+) calcd for C20H22ClO3 (MH+) 345.1257, found 345.1259.3-(4-Bromophenyl)-6,6-dimethyl-2-(3-oxobutyl)-6,7-dihydro-benzofuran-4(5H)-one (12h)
Brown solid; yield 125 mg, 35%; mp 109–110 °C; IR (KBr, cm−1) 1716 (s), 1677 (vs.), 758 (m); 1H NMR (CDCl3, 500 MHz) δ 7.51 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 2.92 (t, J = 7.4 Hz, 2H), 2.79 (t, J = 7.4 Hz, 2H), 2.75 (s, 2H), 2.37 (s, 2H), 2.16 (s, 3H), 1.17 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 20.4, 28.7, 30.0, 35.1, 37.8, 41.5, 53.1, 118.5, 118.6, 121.6, 130.4, 131.3, 131.6, 151.4, 165.4, 193.7, 206.9; MS (ES+) m/z (rel intensity) 429 (MK + 2]+, 90), 427 (MK+, 91), 413 ([MNa + 2]+, 98), 411 (MNa+, 100), 129 (12); HRMS (ES+) calcd for C20H21O3BrNa (MNa+) 411.0566, found 411.0570.6,6-Dimethyl-2-(3-oxobutyl)-3-(thiophen-2-yl)-6,7-dihydrobenzofuran-4(5H)-one (12i)
Black solid; yield 84 mg, 29%; mp 128–130 °C; IR (KBr, cm−1) 717 (s), 1677 (s); 1H NMR (CDCl3, 400 MHz) δ 7.36 (dd, J = 3.5 Hz, J = 1.1 Hz, 1H), 7.30 (dd, J = 5.2 Hz, J = 1.1 Hz, 1H), 7.05 (dd, J = 5.2 Hz, J = 3.5 Hz, 1H), 3.07 (t, J = 7.2 Hz, 2H), 2.81 (t, J = 7.2 Hz, 2H), 2.72 (s, 2H), 2.38 (s, 2H), 2.16 (s, 3H), 1.15 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 21.2, 28.7, 30.0, 34.9, 37.8, 41.7, 53.2, 113.1, 118.6, 125.4, 127.2, 128.6, 131.9, 151.9, 165.3, 193.4, 207.0; MS (ES+) m/z (rel intensity) 317 (MH+, 84), 259 (100); HRMS (ES+) calcd for C18H21O3S (MH+) 317.1211, found 317.1218.6,6-Dimethyl-3-(naphthalen-1-yl)-2-(3-oxobutyl)-6,7-dihydro-benzofuran-4(5H)-one (12j)
Brown solid; yield 129 mg, 39%; mp 116–118 °C; IR (KBr, cm−1) 1717 (s), 1679 (vs.); 1H NMR (CDCl3, 400 MHz) δ 7.86 (t, J = 7.2 Hz, 2H), 7.59 (d, J = 8.5 Hz, 1H), 7.52–7.35 (m, 4H), 2.85, 2.80 (AB, J = 16.0 Hz, 2H), 2.78–2.71 (m, 2H), 2.70–2.62 (m, 2H), 2.45, 2.33 (AB, J = 16.0 Hz, 2H), 2.00 (s, 3H), 1.21 (s, 3H), 1.18 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 20.6, 28.8, 28.8, 29.8, 35.2, 37.8, 41.6, 52.7, 117.1, 120.4, 125.3, 125.7, 125.8, 126.0, 127.9, 128.3, 128.4, 129.5, 132.7, 133.6, 152.2, 164.9, 192.9, 206.9; MS (ES+) m/z (rel intensity) 399 (MK+, 399), 383 (MNa+, 100), 361 (20), HRMS (ES+) calcd for C24H24O3Na (MNa+) 383.1618, found 383.1617.2-(3-Oxobutyl)-3-phenyl-6,7-dihydrobenzofuran-4(5H)-one (13a)
Dark brown solid; yield 86 mg, 33%; mp 63–65 °C; IR (KBr, cm−1) 1716 (s), 1676 (vs.); 1H NMR (CDCl3, 500 MHz) δ 7.38–7.28 (m, 5H), 2.92 (t, J = 7.5 Hz, 2H), 2.86 (t, J = 6.4 Hz, 2H), 2.76 (t, J = 7.5 Hz, 2H), 2.46 (t, J = 6.4 Hz, 2H), 2.16 (quint, J = 6.4 Hz, 2H), 2.12 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 20.4, 22.6, 23.8, 29.9, 38.7, 41.7, 119.7, 119.9, 127.4, 128.1, 129.9, 131.5, 150.9, 166.1, 194.1, 207.0; HRMS (ES+) calcd for C18H18O3Na (MNa+) 305.1148 found 305.1150.2-(3-Oxobutyl)-3-p-tolyl-6-7-dihydrobenzenefuran-4(5H)-one (13b)
Brown solid; yield 125 mg, 46%; mp 91–92 °C; IR (KBr, cm−1) 1716 (s), 1677 (s); 1H NMR (CDCl3, 400 MHz) δ 7.25, 7.18 (AB, J = 8.0 Hz, 4H), 2.91 (t, J = 7.3 Hz, 2H), 2.86 (t, J = 6.2 Hz, 2H), 2.76 (t, J = 7.3 Hz, 2H), 2.46 (t, J = 6.2 Hz, 2H), 2.36 (s, 3H), 2.16 (quint, J = 6.2 Hz, 2H), 2.12 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 20.4, 21.4, 22.6, 23.8, 30.0, 38.8, 41.8, 119.6, 120.0, 128.4, 128.9, 129.7, 137.1, 150.7, 166.0, 194.1, 207.1; MS (ES+) m/z (rel intensity) 297 (MH+, 90), 239 (100), 204 (5), 200 (7); HRMS (ES+) calcd for C19H21O3 (MH+) 297.1491, found 297.1493.3-(3-Methoxyphenyl)-2-(3-oxobutyl)-6,7-dihydrobenzofuran-4(5H)-one (13c)
Brown solid; yield 109 mg, 38%; mp 117–119 °C; IR (KBr, cm−1) 1716 (s), 1676 (vs.); 1H NMR (CDCl3, 500 MHz) δ 7.27 (t, J = 7.9 Hz, 1H), 6.94 (dd, J = 2.4, 1.5 Hz, 1H), 6.91 (dd, J = 7.9, 1.5 Hz, 1H), 6.85 (dd, J = 7.9, 2.4 Hz, 1H), 3.81 (s, 3H), 2.93 (t, J = 7.5 Hz, 2H), 2.86 (t, J = 6.4 Hz, 2H), 2.76 (t, J = 7.5 Hz, 2H), 2.46 (t, J = 6.4 Hz, 2H), 2.16 (quint, J = 6.4 Hz, 2H), 2.12 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 20.5, 22.6, 23.9, 30.0, 38.8, 41.7, 55.4, 113.3, 115.6, 119.6, 119.9, 122.3, 129.0, 132.8, 151.0, 159.3, 166.1, 194.0, 207.0; MS (ES+) m/z (rel intensity) 351 (MK+, 98), 335 (MNa+, 100); HRMS (ES+) calcd for C19H20O4Na (MNa+) 335.1254, found 335.1253.3-(4-Methoxyphenyl)-2-(3-oxobutyl)-6,7-dihydrobenzofuran-4(5H)-one (13d)
Yellow solid; yield 132 mg, 46%; mp 136–137 °C; IR (KBr, cm−1) 1716 (m), 1673 (s); 1H NMR (CDCl3, 400 MHz) δ 7.29 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 3.81 (s, 3H), 2.90 (t, J = 7.8 Hz, 2H), 2.86 (t, J = 6.3 Hz, 2H), 2.76 (t, J = 7.8 Hz, 2H), 2.46 (t, J = 6.3 Hz, 2H), 2.17 (quint, J = 6.3 Hz, 2H), 2.12 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 20.4, 22.6, 23.9, 30.0, 38.8, 41.8, 55.4, 113.6, 119.3, 120.0, 123.7, 131.0, 150.5, 158.9, 166.0, 194.3, 207.1; MS (ES+) m/z (rel intensity) 335 (MNa+, 50), 313 (MH+, 100), 256 (18), 255 (96); HRMS (ES+) calcd for C19H21O4 (MH+) 313.1440, found 313.1437.3-(3,4-Dimethoxyphenyl)-2-(3-oxobutyl)-6,7-dihydrobenzofuran-4(5H)-one (13e)
Yellow solid; yield 94 mg, 30%; mp 113–114 °C; IR (KBr, cm−1) 1716 (s), 1674 (vs.); 1H NMR (CDCl3, 400 MHz) δ 7.00 (s, 1H), 6.90–6.88 (unresolved AB, 2H), 3.90 (s, 6H), 2.95 (t, J = 7.0 Hz, 2H), 2.88 (t, J = 6.1 Hz, 2H), 2.78 (t, J = 7.0 Hz, 2H), 2.48 (t, J = 6.1 Hz, 2H), 2.18 (quint, J = 7.0 Hz, 2H), 2.14 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 20.5, 22.6, 23.9, 30.0, 38.8, 41.7, 56.0, 56.1, 110.8, 113.7, 119.5, 119.9, 122.0, 124.0, 148.4, 150.6, 166.1, 194.3, 207.1; MS (ES+) m/z (rel intensity) 365 (MNa+, 60), 343 (MH+, 88), 286 (21), 285 (100); HRMS (ES+) calcd for C20H23O5 (MH+) 343.1545, found 343.1545.3-(4-Bromophenyl)-2-(3-oxobutyl)-6,7-dihydrobenzofuran-4(5H)-one (13f)
Brown solid; yield 139 mg, 42%; mp 90–92 °C; IR (KBr, cm−1) 1717 (s), 1676 (vs.), 762 (m); 1H NMR (CDCl3, 500 MHz) δ 7.49 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 2.90 (t, J = 7.4 Hz, 2H), 2.87 (t, J = 6.4 Hz, 2H), 2.76 (t, J = 7.4 Hz, 2H), 2.47 (t, J = 6.4 Hz, 2H), 2.17 (quint, J = 6.4 Hz, 2H), 2.13 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 20.2, 22.4, 23.7, 29.9, 38.5, 41.4, 118.6, 119.6, 121.5, 130.3, 131.1, 131.4, 150.9, 166.1, 194.0, 206.7; MS (ES+) m/z (rel intensity) 385 ([M + 2]Na+, 100), 383 (MNa+, 98); HRMS (ES+) calcd for C18H17O3BrNa (MNa+) 383.0253 found 383.0253.2-(3-Oxobutyl)-3-(thiophen-2-yl)-6,7-dihydrobenzofuran-4(5H)-one (13g)
Black solid; yield 40 mg, 15%; mp 104–105 °C; IR (KBr, cm−1) 1715 (s), 1676 (s); 1H NMR (CDCl3, 400 MHz) δ 7.31–7.29 (m, 2H), 7.05 (dd, J = 5.1, 3.6 Hz, 1H), 3.04 (t, J = 6.9 Hz, 2H), 2.85 (t, J = 6.1 Hz, 2H), 2.79 (t, J = 6.9 Hz, 2H), 2.48 (t, J = 6.1 Hz, 2H), 2.21–2.14 (m, 2H), 2.15 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 21.0, 22.4, 23.8, 30.0, 38.8, 41.7, 113.1, 119.8, 125.5, 127.2, 128.5, 131.9, 151.7, 166.1, 194.0, 207.0; MS (ES+) m/z (rel intensity) 289 (MH+, 70), 233 (5), 232 (15), 231 (100), 220; HRMS (ES+) calcd for C16H17O3S (MH+) 289.0898, found 289.0887.3-(Naphthalen-1-yl)-2-(3-oxobutyl)-6,7-dihydrobenzofuran-4(5H)-one (13h)
Dark brown solid; yield 134 mg, 44%; mp 91–93 °C; IR (KBr, cm−1) 1715 (m), 1678 (vs.); 1H NMR (CDCl3, 500 MHz) δ 7.85 (t, J = 8.2 Hz, 2H), 7.59 (d, J = 8.2 Hz, 1H), 7.49–7.33 (m, 4H), 2.95 (AB, J = 6.4 Hz, 2H), 2.78–2.70 (m, 2H), 2.68–2.58 (m, 2H), 2.50–2.38 (m, 2H), 2.24–2.18 (m, 2H), 2.00 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 20.6, 22.7, 23.9, 29.8, 38.5, 41.6, 117.3, 121.7, 125.4, 125.8, 125.8, 126.1, 127.9, 128.4, 128.5, 129.6, 132.7, 133.7, 151.9, 165.8, 193.7, 207.0; MS (ES+) m/z (rel intensity) 371 (MK+, 12), 355 (MNa+, 100), 305 (4); HRMS (ES+) calcd for C22H20O3Na (MNa+) 355.1305, found 355.1303.Acknowledgements
INNN thanks DST India for financial assistance. VM and TK thank CSIR India for a research fellowship.References
- (a) A. Williams, Chemical Technology Review, No. 18: Furans: Synthesis and Applications, Noyes Data Corp., Park Ridge, N. J., 1973, p. 303 Search PubMed; (b) B. A. Keay and P. W. Dibble, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Sciven, Elsevier, Oxford, 1997, vol. 2, p. 395 Search PubMed; (c) D. M. X. Donnelly and M. J. Meegan, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky and C. W. Rees, Pergamon, Oxford, 1984, vol. 4, p. 657 Search PubMed; (d) B. H. Lipshutz, Chem. Rev., 1986, 86, 795 CrossRef CAS.
- Selected reviews: (a) T. Shibamoto, L. Yu, S. Wang and B.-G. Sun, Food Saf. Chem., 2015, 35 CAS; (b) A. Verma, S. N. Pandeya and S. Sinha, Int. J. Res. Ayurveda Pharm., 2011, 2, 1110 CAS; (c) F. Piozzi, M. Bruno, S. Rosselli and A. Maggio, Heterocycles, 2007, 74, 31 CrossRef CAS; (d) Y. Liu, S. Zhang, J. H. Jung and T. Xu, Top. Heterocycl. Chem., 2007, 11, 231 CAS; (e) Y. Liu, A. Zhang and J. M. Pedro, Nat. Prod. Rep., 2006, 23, 630 RSC; (f) H. Ekiert and K. G. Ramawat, Biotechnol. Med. Plants, 2004, 267 CAS; (g) M. Taniguchi and K. Baba, Foods Food Ingredients J. Jpn., 2002, 203, 34 CAS; (h) E. Abele and E. Lukevics, Chem. Heterocycl. Compd., 2001, 37, 141 CrossRef CAS; (i) D. S. Brown and L. A. Paquette, Heterocycles, 1992, 34, 807 CrossRef CAS; Selected articles: (j) Anti-microbial: N. Zanatta, S. H. Alves, H. S. Coelho, D. M. Borchhardt, P. Machado, K. M. Flores, F. M. Silva, T. B. Spader, J. M. Santurio, H. G. Bonacorso and M. A. P. Martins, Bioorg. Med. Chem., 2007, 15, 1947 CrossRef CAS PubMed; (k) M. W. Khan, M. J. Alam, M. A. Rashid and R. Chowdhury, Bioorg. Med. Chem., 2005, 13, 4796 CrossRef CAS PubMed; (l) Anticancer: R. R. Rao, V. Chaturvedi, K. S. Babu, P. P. Reddy, V. R. S. Rao, P. Sreekanth, A. S. Sreedhar and J. M. Rao, Med. Chem. Res., 2012, 21, 634 CrossRef CAS; (m) Tubulin polymerization inhibitor: D. J. Kerr, E. Hamel, M. K. Jung and B. L. Flynn, Bioorg. Med. Chem., 2007, 15, 3290 CrossRef CAS PubMed; (n) Antipsoriatic: A. Reichstein, S. Vortherms, S. Bannwitz, J. Tentrop, H. Prinz and K. Muller, J. Med. Chem., 2012, 55, 7273 CrossRef CAS PubMed; (o) Anti-hyperglycemic: M. S. Malamas, J. Sredy, C. Moxham, A. Katz, W. Xu, R. McDevitt, F. O. Adebayo, D. R. Sawicki, L. Seestaller, D. Sullivan and J. R. Taylor, J. Med. Chem., 2000, 43, 1293 CrossRef CAS PubMed; (p) Anti-HIV agent: C. W. Chang and R. E. Chein, J. Org. Chem., 2011, 76, 4154 CrossRef CAS PubMed; (q) Anti-tuberculosis: R. P. Tangallapally, R. Yendapally, R. E. Lee, A. J. M. Lenaerts and E. Lee, J. Med. Chem., 2005, 48, 8261 CrossRef CAS PubMed.
- Selected reviews: (a) R. C. D. Brown, Angew. Chem., Int. Ed., 2005, 44, 850 CrossRef CAS PubMed; (b) X. L. Hou, H. Y. Cheung, T. Y. Hon, P. L. Kwan, T. H. Lo, S. Y. Tong and H. N. C. Wong, Tetrahedron, 1998, 54, 1955 CrossRef CAS; (c) S. F. Kirsch, Org. Biomol. Chem., 2006, 4, 2076 RSC; (d) F. W. Lewis and L. M. Harwood, Targets Heterocycl. Syst., 2012, 16, 1 CAS; (e) S. J. Sabounchei, F. A. Bagherjeri and M. Panahimehr, Adv. Chem. Res., 2012, 15, 31 CAS; (f) B. Alicia and L. Alvarez, Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, 2011, p. 99 Search PubMed; (g) A. Gandini, Biopolymers, 2011, 179 CAS; (h) I. B. Masesane, Trends Org. Chem., 2010, 14, 13 CAS; (i) A. Gandini, Green Polym. Methods, 2011, 29 CAS.
- (a) H. Kwiecien, M. Smist and M. Kowalewska, Curr. Org. Synth., 2012, 9, 529 CrossRef CAS; (b) M. G. Uchuskin, V. A. Shcherbinin and A. V. Butin, Chem. Heterocycl. Compd., 2014, 50, 619 CrossRef CAS; (c) T. T. Haug and S. F. Kirsch, Targets Heterocycl. Syst., 2009, 13, 57 CAS.
- Review: (a) S. Khaghaninejad and M. M. Heravi, Adv. Heterocycl. Chem., 2014, 111, 95 CrossRef CAS PubMed; articles: (b) V. Amarnath and K. Amarnath, J. Org. Chem., 1995, 60, 301 CrossRef CAS; (c) G. Minetto, L. F. Raveglia, A. Sega and M. Taddei, Eur. J. Org. Chem., 2005, 5277 CrossRef CAS PubMed.
- (a) F. Feist, Chem. Ber., 1902, 35, 1537 CrossRef CAS PubMed; (b) E. Benary, Chem. Ber., 1911, 44, 489 CrossRef CAS PubMed; (c) G. Mross, E. Holtz and P. Langer, J. Org. Chem., 2006, 71, 8045 CrossRef CAS PubMed; (d) N. Ayyagari, D. Jose, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2011, 52, 258 CrossRef CAS PubMed.
- Selected reviews: (a) W. J. Moran and A. Rodriguez, Org. Prep. Proced. Int., 2012, 44, 103 CrossRef CAS PubMed; (b) A. S. Dudnik and V. Gevorgyan, Catalyzed Carbon-Heteroatom Bond Formation, ed. A. K. Yudin, 2011, p. 317 Search PubMed; (c) N. T. Patil and Y. Yamamoto, ARKIVOC, 2007, 10, 121 CrossRef; Selected articles: (d) A. S. K. Hashmi, L. Schwarz, J. H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285 CrossRef CAS; (e) S. Ma, J. Zhang and L. Lu, Chem.–Eur. J., 2003, 9, 2447 CrossRef CAS PubMed; (f) J. A. Marshall and G. S. Bartley, J. Org. Chem., 1994, 59, 7169 CrossRef CAS; (g) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 5195 CrossRef CAS PubMed; (h) A. G. M. Barrett, Chem. Soc. Rev., 1991, 20, 95 RSC.
- Selected recent reviews: (a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed; (b) D. Basavaiah and B. C. Sahu, Chimia, 2013, 67, 8 CrossRef CAS PubMed; (c) G. Guillena, D. J. Ramon and M. Yus, Catalysis, 2012, 24, 223 CAS; (d) D. Basavaiah and G. Veeraraghavaiah, Chem. Soc. Rev., 2012, 41, 68 RSC; (e) V. Singh and S. Batra, Tetrahedron, 2008, 64, 4511 CrossRef CAS PubMed.
- (a) C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069 CrossRef CAS PubMed and the references cited therein; (b) P. Xie and Y. Huang, Eur. J. Org. Chem., 2013, 6213 CrossRef CAS PubMed; see also: (c) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS PubMed.
- (a) Review: K. Kaur and I. N. N. Namboothiri, Chimia, 2012, 66, 913 CrossRef CAS PubMed; selected articles: (b) V. Barbier, F. Couty and O. R. P. David, Eur. J. Org. Chem., 2015, 3679 CrossRef CAS PubMed; (c) B. Han, X. Xie, W. Huang, X. Li, L. Yang and C. Peng, Adv. Synth. Catal., 2014, 356, 3676 CrossRef CAS PubMed; (d) J.-Q. Zhang, J.-J. Liu, C.-L. Gu, D. Wang and L. Liu, Eur. J. Org. Chem., 2014, 5885 CrossRef CAS PubMed; (e) N. Lu, H. Wang and Y. Wang, Bull. Korean Chem. Soc., 2013, 34, 3591 CrossRef CAS; (f) X.-Y. Chen, F. Xia and S. Ye, Org. Biomol. Chem., 2013, 11, 5722 RSC; (g) N. Lu, L. Meng, D. Chen and G. Zhang, RSC Adv., 2011, 1, 1113 RSC; (h) X. Wang, Y.-F. Chen, L.-F. Niu and P.-F. Xu, Org. Lett., 2009, 11, 3310 CrossRef CAS PubMed; (i) S. Chandrasekhar, K. Mallikarjun, G. Pavankumarreddy, K. V. Rao and B. Jagadeesh, Chem. Commun., 2009, 4985 RSC; (j) S. Fioravanti, L. Pellacani and P. A. Tardella, Tetrahedron, 2009, 65, 5747 CrossRef CAS PubMed; (k) Y. Wu, A. Lu, Y. Liu, X. Yu, Y. Wang, G. Wu, H. Song, Z. Zhou and C. Tang, Tetrahedron: Asymmetry, 2010, 21, 2988 CrossRef CAS PubMed; (l) Y. Wang, S. Zhu and D. Ma, Org. Lett., 2011, 13, 1602 CrossRef CAS PubMed; (m) A. Rai and L. D. S. Yadav, Tetrahedron, 2012, 68, 2459 CrossRef CAS PubMed; (n) Z.-W. Guo, J.-W. Xie, C. Chen and W.-D. Zhu, Org. Biomol. Chem., 2012, 10, 8471 RSC; (o) G. Talavera, E. M. Reyes, J. L. Vicario and L. Carrillo, Angew. Chem., Int. Ed., 2012, 51, 4104 CrossRef CAS PubMed; (p) B.-C. Hong, D.-J. Lan, N. S. Dange, G.-H. Lee and J.-H. Liao, Eur. J. Org. Chem., 2013, 2472 CrossRef CAS PubMed; (q) J. An, L.-Q. Lu, Q.-Q. Yang, T. Wang and W.-J. Xiao, Org. Lett., 2013, 15, 542 CrossRef CAS PubMed; (r) M. Yaqub, C. Y. Yu, Y. M. Jia and Z. T. Huang, Synlett, 2008, 1357 CAS; (s) S. Takizawa, A. Horii and H. Sasai, Tetrahedron: Asymmetry, 2010, 21, 891 CrossRef CAS PubMed; (t) N. Lu, L. Meng, D. Chen and G. Zhang, RSC Adv., 2011, 1, 1113 RSC; (u) M. Bakthadoss, N. Sivakumar, A. Devaraj and D. S. Sharada, Synthesis, 2011, 2196 Search PubMed; (v) R. Kumar, T. Kumar, S. M. Mobin and I. N. N. Namboothiri, J. Org. Chem., 2013, 78, 5073 CrossRef CAS PubMed.
- (a) Terphenyls: E. Gopi and I. N. N. Namboothiri, J. Org. Chem., 2014, 79, 7468 CrossRef CAS PubMed; (b) Cyclopentenes: L. F. Yeh, S. Anwar and K. Chen, Tetrahedron, 2012, 68, 7317 CrossRef CAS PubMed; (c) Bicyclic skeletons: C.-L. Cao, Y.-Y. Zhou, J. Zhou, X.-L. Sun, Y. Tang, Y.-X. Li, G.-Y. Li and J. Sun, Chem.–Eur. J., 2009, 15, 11384 CrossRef CAS PubMed.
- Imidazopyridines: (a) D. K. Nair, S. M. Mobin and I. N. N. Namboothiri, Org. Lett., 2012, 14, 4580 CrossRef CAS PubMed; (b) Pyranonaphthaquinones: D. K. Nair, R. F. S. Menna-Barreto, E. N. da Silva Júnior, S. M. Mobin and I. N. N. Namboothiri, Chem. Commun., 2014, 50, 6973 RSC; (c) Imidazoles: T. Kumar, D. Verma, R. F. S. Menna-Barreto, W. O. Valenca, E. N. da Silva Júnior and I. N. N. Namboothiri, Org. Biomol. Chem., 2015, 13, 1996 RSC; pyrroles: (d) D. R. Magar, Y.-J. Ke and K. Chen, Asian J. Org. Chem., 2013, 2, 330 CrossRef CAS PubMed; (e) T. Chen, N. Shao, H. Zhu, B. Zhang and H. Zou, Tetrahedron, 2013, 69, 10558 CrossRef CAS PubMed; (f) Oxa- and aza-triquinanes: J. An, L.-Q. Lu, Q.-Q. Yang, T. Wang and W.-J. Xiao, Org. Lett., 2013, 15, 542 CrossRef CAS PubMed; (g) Tetrahydropyridines: M. Yaqub, C.-Y. Yu, Y.-M. Jia and Z.-T. Huang, Synlett, 2008, 1357 CAS; (h) Different heterocyclic scaffolds: H. Zhu, N. Shao, T. Chen and H. Zou, Chem. Commun., 2013, 49, 7738 RSC.
- Furans from 1,3-dicarbonyl compounds: (a) D. K. Nair, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2012, 53, 3349–3352 CrossRef CAS PubMed; (b) W.-Y. Huang, Y.-C. Chen and K. Chen, Chem.–Asian J., 2012, 7, 688 CrossRef CAS PubMed; arenofurans from arenols: (c) T. Kumar, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron, 2013, 69, 4964 CrossRef CAS PubMed; (d) S. Anwar, W.-Y. Huang, C.-H. Chen, Y.-S. Cheng and K. Chen, Chem.–Eur. J., 2013, 19, 4344 CrossRef CAS PubMed.
- (a) M. Dadwal, R. Mohan, D. Panda, S. M. Mobin and I. N. N. Namboothiri, Chem. Commun., 2006, 338 RSC; (b) P. Shanbhag, P. R. Nareddy, M. Dadwal, S. M. Mobin and I. N. N. Namboothiri, Org. Biomol. Chem., 2010, 8, 4867 RSC; (c) See also ref. 10.
- (a) R. Muruganantham and I. N. N. Namboothiri, J. Org. Chem., 2010, 75, 2197 CrossRef CAS PubMed; (b) R. Kumar and I. N. N. Namboothiri, Org. Lett., 2011, 13, 4016 CrossRef CAS PubMed; (c) D. Verma, R. Kumar and I. N. N. Namboothiri, J. Org. Chem., 2013, 78, 3482 CrossRef CAS PubMed.
- P. Shanbhag, V. Mane, C. Hazra and I. N. N. Namboothiri, J. Indian Chem. Soc., 2013, 90, 1713 CAS.
- (a) Ref. 14b; (b) C. R. Reddy, M. D. Reddy and K. Haribabu, Eur. J. Org. Chem., 2012, 6414 CrossRef CAS PubMed.
- S. Roy, M. Amireddy and K. Chen, Tetrahedron, 2013, 69, 8751 CrossRef CAS PubMed.
- X. Huang, H. Shi, J. Ren, G. Liu, Y. Tang and B. Zeng, Chin. J. Chem., 2012, 30, 1305 CrossRef CAS PubMed.
- The reaction of compound 1 with cyclopentane-1,3-dione provides fused pyran, see ref. 13a and b.
- See for similar mechanism in the formation of pyrrole and furan from nitroalkenes: L. Li, M.-N. Zhao, Z.-H. Ren, J. Li and Z.-H. Guan, Synthesis, 2012, 532 CAS.
- (a) I. Deb, M. Dadwal, S. M. Mobin and I. N. N. Namboothiri, Org. Lett., 2006, 8, 1201 CrossRef CAS PubMed; (b) I. Deb, P. Shanbhag, S. M. Mobin and I. N. N. Namboothiri, Eur. J. Org. Chem., 2009, 4091 CrossRef CAS PubMed; (c) R. J. Reddy and K. Chen, Org. Lett., 2011, 13, 1458 CrossRef CAS PubMed.
- J. J. Lee, J. Kim, Y. M. Jun, B. M. Lee and B. H. Kim, Tetrahedron, 2009, 65, 8821 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies NMR spectra. See DOI: 10.1039/c5ra11471c |
| This journal is © The Royal Society of Chemistry 2015 |