
Self-healing elastomer assembly towards three-dimensional shape memory devices†
Abstract
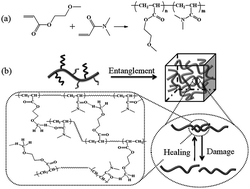
The superior self-healing elastomers were readily prepared by free-radical copolymerization of methoxyethyl acrylate (MEA) and N,N-dimethylacrylamide (DMAA). The synergistic interaction between the shape memory effect and the reversible weak hydrogen bonds lead to the excellent self-healing properties and high mechanical strength of the elastomers. By taking advantage of the two outstanding performances, we demonstrated that various novel two-dimensional (2D) and three-dimensional (3D) shape memory devices can be precisely designed on the basis of the self-healing mediated assembly of the elastomers as building blocks. This assembly method to combine self-healing and shape memory properties might open up a promising avenue for the design and fabrication of 3D complex-shape smart devices.
 Please wait while we load your content...
Please wait while we load your content...

