
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water soluble hyperbranched polymers from controlled radical homopolymerization of PEG diacrylate†
Tianyu
Zhao
a,
Hong
Zhang
a,
Dezhong
Zhou
a,
Yongsheng
Gao
a,
Yixiao
Dong
b,
Udo
Greiser
a,
Hongyun
Tai
c and
Wenxin
Wang
*ad
aCharles Institute of Dermatology, School of Medicine and Medical Science, University College Dublin, Dublin, Ireland. E-mail: wenxin.wang@ucd.ie; Tel: +353 01 7166341
bSchool of Medicine, Stanford University, 291 Campus Drive Li Ka Shing Building, Stanford, USA
cSchool of Chemistry, Bangor University, Bangor, LL57 2UW, UK
dSchool of Chemistry and Chemical Biology, University College Dublin, Dublin, Ireland
First published on 7th April 2015
Abstract
A series of water soluble PEG based hyperbranched polymers were successfully synthesized by homopolymerization of poly(ethylene glycol) diacrylate (PEGDA) (Mn = 575 and 700 g mol−1 respectively) via vinyl oligomer combination. The homopolymerization of diacrylate macromers underwent a slow vinyl propagation combined with a polycondensation by coupling of reactive oligomers. At a high initiator-to-monomer ratio (e.g. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), high monomer conversions up to 96% were achieved in concentrated reaction conditions (60% w/v) without gelation. The hyperbranched polymers obtained from homopolymerization of PEGDA575 show concentration-dependent thermoresponsive properties in aqueous solutions.
:2), high monomer conversions up to 96% were achieved in concentrated reaction conditions (60% w/v) without gelation. The hyperbranched polymers obtained from homopolymerization of PEGDA575 show concentration-dependent thermoresponsive properties in aqueous solutions.
Introduction
Controlled radical polymerization (CRP) of multivinyl monomers (MVMs) has attracted considerable attention as it provides possibilities to synthesize polymers with controlled 3D macromolecule structures, molecular weights, degree of branching and crosslinking density. Since the “Strathclyde synthesis” was introduced for the preparation of branched polymers,1–6 the majority of polymerizations involving MVMs have been carried out in copolymerization systems containing a low percentage of MVMs. Pioneering studies have been published by Armes's group7–14 and Matyjaszewski's group15–19 reporting branched polymers with long linear primary chains linked together. Until recently, attention has been focused on the homopolymerization of MVMs via CRP to produce polymers with linear,20 branched21–23 or cyclic structures.24–26 Homopolymerization had not been widely explored previously because it is generally accepted that the critical gelation happens when the average number of crosslinkages (crosslinker in which both vinyl groups have reacted) per primary chain exceeds unity if the primary chains are uniform,16 according to Flory–Stockmayor's mean-field theory. However, it has been shown by many experiments that if the monomer conversion is kept incomplete and a portion of the divinyl cross-linker does not fully react, or is consumed by intramolecular cyclization, gelation could be avoided. These results provide us with two classical alternative polymer products, i.e. either highly branched structure (no cyclization) at a low conversion or cyclized products at a high conversion. Most importantly, it has been realized by an increasing number of researchers that intramolecular cyclization cannot be neglected in the CRP of MVMs, including the homopolymerization and copolymerization.27–30 The intramolecular cyclization could be useful to create novel macromolecular structures, such as knots24,25 or ladders.31,32 However, it consumes the vinyl functional groups on the polymer chains and adds difficulties to the syntheses of purely branched structures. Although many asymmetrical divinyl monomers33–35 were successfully employed for the syntheses of hyperbranched polymers with pendant vinyl groups, this approach suffers from its lack of versatility as it implies tailor-synthesized asymmetrical divinyl monomers.To avoid the ‘loops’ formed by the intramolecular cyclization, it is conceivable that an increase in polymer chain concentration would favor the intermolecular branching. The critical overlap concentration, c*, has been used to predict the dominant interactions in the MVM system.10,24,36 A concentration below c* implies the dominant interactions are intramolecular and a value above c* implies more intermolecular branching. However, if chain concentration is raised by reducing solvent proportion, the kinetic control for the CRP could recede and gelation could occur in advance. Thus, we sought to find another approach which is based on the increase of the initiator concentration to quickly increase polymer chain concentration. The advantages of high initiator concentration are: firstly, the high ratio of initiator to divinyl monomer can lead to extremely short primary chains and thus decrease the chance of primary intramolecular cyclization at the early stage of reaction; secondly, the concurrent chain growth could create a rapid increase in chain concentration for enhanced intermolecular branching and thirdly, the high ratio of initiator to divinyl monomer can delay the gelling point according to Flory–Stockmayer's statistical theory17 in order to achieve a high monomer conversion. Therefore, a new strategy ‘vinyl oligomer combination’ has been developed for the preparation of hyperbranched polymers through CRP of MVMs. In this strategy, linear oligomers with pendant vinyl groups are formed by the slow chain growth of divinyl monomers at an early stage and as the reaction progresses these linear oligomer chains combine through the pendant vinyl groups of other linear oligomers to form highly branched polymers.
In this study, two poly(ethylene glycol) diacrylates (PEGDA) (PEGDA575, average Mn = 575; PEGDA700 average Mn = 700) having different lengths of PEG spacers (n = 10 and 13 respectively, as shown in Scheme 1) were homopolymerized in a concentrated solution ([PEGDA] = 60% w/v) via in situ deactivation enhanced ATRP (DE-ATRP) to produce a series of water-soluble hyperbranched polymers. The relative propensities for intermolecular propagating/cross-linking reactions and intramolecular cyclization were assessed using gel permeation chromatography (GPC)/viscometer and 1H NMR measurements. Finally, the concentration-dependent phase transition behaviors of the obtained poly(PEGDA575)s were evaluated.
 | ||
| Scheme 1 Homopolymerization of PEGDA through vinyl oligomer combination. | ||
Experimental section
Materials
Poly(ethylene glycol) diacrylate (PEGDA575 and PEGDA700, 98% Sigma-Aldrich), tert-butyl α-bromoisobutyrate (BBriB, 98%, Aldrich), pentamethyldiethylenetriamine (PMDETA, 99%, Aldrich), copper(II) chloride (CuCl2, 97%, Aldrich), L-ascorbic acid (AA, 99%, Aldrich), d-chloroform (99.8%, Aldrich), 2-butanone (HPLC grade, Aldrich), chloroform (HPLC grade, Aldrich) and diethyl ether (ACS reagent grade, Aldrich) were used as received.Synthesis of hyperbranched poly(PEGDA)
The polymers were prepared in two-neck round bottom flasks. 2-Butanone, CuCl2 (67.3 mg, 1 equiv.) and PMDETA (173 mg, 2 equiv.) were added into the flask, following the addition of the required amount of PEGDA monomer and initiator. The initiator-to-monomer ratios used are stated in Table 1, which are 1:2, 1:4 and 1:8 for both poly(PEGDA575) and poly(PEGDA700). Oxygen was removed by bubbling argon through the solutions for 20 min. AA solution (173 μl of 0.1 mg μl−1 AA/H2O solution, 0.2 equiv.) was pipetted into the flasks under positive pressure of argon before the flask was immersed in a pre-heated oil bath at 50 °C. The solution was stirred at 800 rpm and the polymerization was conducted at 50 °C in an oil bath for the required reaction time. The experiment was stopped by opening the flask and exposing the catalyst to air. The solution was then diluted with THF and precipitated into a large excess of cold diethyl ether to remove the monomers. The precipitated mixture was dried under laminar flow then re-dissolved in acetone, followed by three times of passing through an Al2O3 column. The mixture was then dried under vacuum.
| Entry | Diacrylate | I:Ma |
Time (h) | Monomer conv.b (%) | Yieldc (%) | M w (Mw/Mn) (kg mol−1) | Vinyl contente (%) | Branch ratioe (%) | Initiator contentf (%) | α g | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GPC-RId | GPC-viscod | ||||||||||
| a [M]/[I]/[CuCl2]/[PMDETA]/[AA] = 80/[I](=40; 20; 10)/1/2/0.2, M: polyethylene glycol diacrylate, I: tert-butyl α-bromoisobutyrate (BBriB), PMDETA: 1,1,4,7,7-pentamethyldiethylenetriamine, AA: L-ascorbic acid, solvent: 2-butanone. b Monomer conversion is determined by the integration of polymer and monomer peaks in the GPC-RI trace. c Diethyl ether-insoluble part. d M n, Mw are determined by GPC equipped with triple detectors using PMMA as standards in chloroform. e Calculated by 1H NMR as seen in Fig. 3 and eqn (S1) and (S2). f Mole ratio of initiator/PEGDA unit, calculated by 1H NMR and eqn (S3). g Mark–Houwink exponent α. | |||||||||||
| 1 | PEGDA575 | 1:2 |
4.5 | 95.6 | 68 | 403 (6.6) | 531 (16.1) | 37.3 | 62.7 | 41.1 | 0.40 |
| 2 | 1:4 |
6.0 | 94.1 | 65 | 153 (5.6) | 279 (14.5) | 52.7 | 47.3 | 27.8 | 0.34 | |
| 3 | 1:8 |
6.0 | 76.9 | 54 | 94 (4.3) | 177 (11.7) | 61.8 | 38.2 | 19.0 | 0.32 | |
| 4 | PEGDA700 | 1:2 |
4.5 | 90.5 | 45 | 53 (2.5) | 71 (4.9) | 40.7 | 59.3 | 44.2 | 0.35 |
| 5 | 1:4 |
6.0 | 93.2 | 50 | 132 (8.8) | 184 (11.1) | 56.6 | 43.4 | 29.5 | 0.41 | |
| 6 | 1:8 |
6.0 | 70.9 | 46 | 45 (3.2) | 65 (3.8) | 67.6 | 32.4 | 21.5 | 0.33 | |
Molecular weight determination by gel permeation chromatography (GPC)
Samples were taken from the reaction mixture at specific time intervals using a glass syringe with Luer needle under positive pressure of argon. These aliquots were then diluted in chloroform and filtered through an Al2O3 pipette for chromatography followed by a 0.4 μm filter before analysis. The molecular weight and molecular weight distribution of each sample was determined using a GPC PL-50 (Agilent) instrument with triple detectors (RI, viscometer and LS). Chromatography was performed with tow sequential columns (30 cm PL gel Mixed-C columns) at 40 °C using chloroform as eluent with a flow rate of 1 ml min−1. The RI and viscosity detector were calibrated with a series of 12 near-monodisperse PMMA standards (Mp from 690 to 1944000 g mol−1, Agilent).
Nuclear magnetic resonance (NMR) spectroscopy
The polymers were dissolved in CDCl3 for 1H NMR analysis. 1H NMR analysis was carried out on a S4 300 MHz Bruker NMR with MestReNova processing software. The chemical shifts were referenced to the lock chloroform (7.26 ppm). The 1H NMR spectrum confirmed the presence of each monomer in the polymer structure and the presence of free vinyl groups.Phase transition temperature measurement
The phase transition temperatures were determined in water by turbidity measurement on a temperature-controlled UV-vis spectrometer. The light transmittance of polymer aqueous solutions of different concentrations was measured at 500 nm. The phase transition temperatures were defined as the temperature corresponding to 90% transmittance of aqueous solution during the heating process.Results and discussions
To facilitate the intermolecular branching, the molar ratios of initiator to divinyl monomer were set as 1:2, 1:4 and 1:8 for both PEGDA575 and PEGDA700. The reaction conditions and the characterization data of the polymer products were summarized in Tables 1 and S1.† It is known that in ATRP reaction, a high initiator concentration will lead to a shift in equilibrium towards the active radicals. And this usually results in a high concentration of radicals which terminate efficiently, leaving an excess of X–Cu(II), known as the persistent radical effect (PRE). To eliminate the termination, large amount of Cu(II) was introduced initially to stabilize the equilibrium and keep the radical concentration low. Therefore, despite of the high initiator concentration, most of the initiators were in their dormant state because of the enhanced deactivation. Meanwhile, 20% of reducing agent and extra amount of ligand was used to (re)generate Cu(I) and thus maintained the reaction rate to an acceptable level. The polymer chain growth was monitored using GPC at regular time intervals during the reaction (Fig. 1). The traces showed similar evolution of molecule growth in the reaction system. Multimodal peaks at the early stage (before 1.5 h) indicate the formation of monoadducts of monomer and initiator as well as a certain portion of oligomers. The appearance of more broadened peaks at later stages suggests that the combination of lower molecular weight oligomers became the dominant reaction pathway. It is likely that at later stages of the reaction, when most of the monomers were already consumed, the reaction conditions were more favorable for statistical branching rather than for linear growth. Living character of the growing chains was demonstrated by a peak shift in the GPC trace from oligomers to larger molecules throughout the reaction, while molecular weights and the polydispersity index increased significantly (Table S1†), indicating the formation of a hyperbranched structure. It can be seen from the GPC trace that the monomer conversion is high (Table 1), especially for the higher ratio of initiator to monomer (1:2 and 1:4). By precipitation in diethyl ether, the monomers and some lower molecular weight polymers were removed, leaving only the highly branched polymers with higher average molecular weight (as seen from the insertion of Fig. 1). Thus, the molecular weight of the final products (Table 1) is significantly higher than that monitored during the reactions (Table S1†). In order to develop a useful method for the effective preparation of branched polymers, it is desirable to find a reaction condition which allows a higher monomer concentration to be used. Our attempts have demonstrated that both high monomer conversion and hyperbranched polymer structure could be obtained by homopolymerization of diacrylates with a high ratio of initiator at a high monomer concentration (60% w/v).
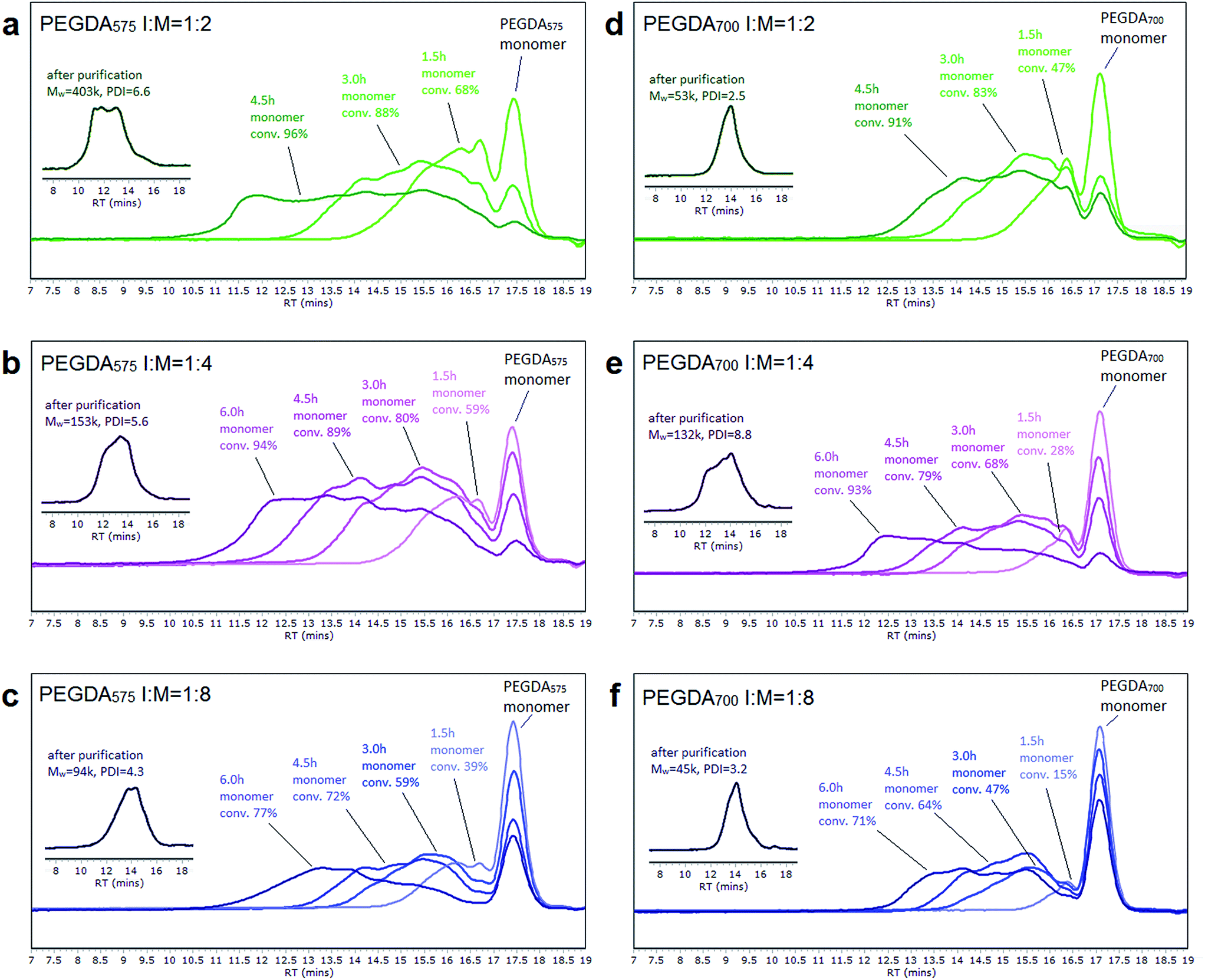 | ||
| Fig. 1 GPC traces of the polymerization mixtures taken at different reaction times for the homopolymerizations of poly(PEGDA)s at different initiator-to-monomer ratios. The inserts are the GPC traces of the final products after purification. | ||
Characterization of the obtained poly(PEGDA)s was conducted by GPC/RI and GPC/viscometer to overcome the erroneous results caused by different hydrodynamic volumes of molecules in different solvents. The weight-average molecular weights for all of the poly(PEGDA)s – as determined by GPC/viscometer – are apparently higher than those obtained from GPC/RI (Table 1), indicating that the products possess highly branched structures rather than linear structures. The Mark–Houwink plots for the obtained poly(PEGDA)s and a linear counterpart (Fig. 2) show that as the molecular weight increases, the viscosity of the obtained poly(PEGDA) solutions increases less than that of the linear poly(PEGMA) synthesized by the same method. The Mark–Houwink exponents of poly(PEGDA)s are significantly low (α = 0.3–0.5), indicating a more compact dense structure.
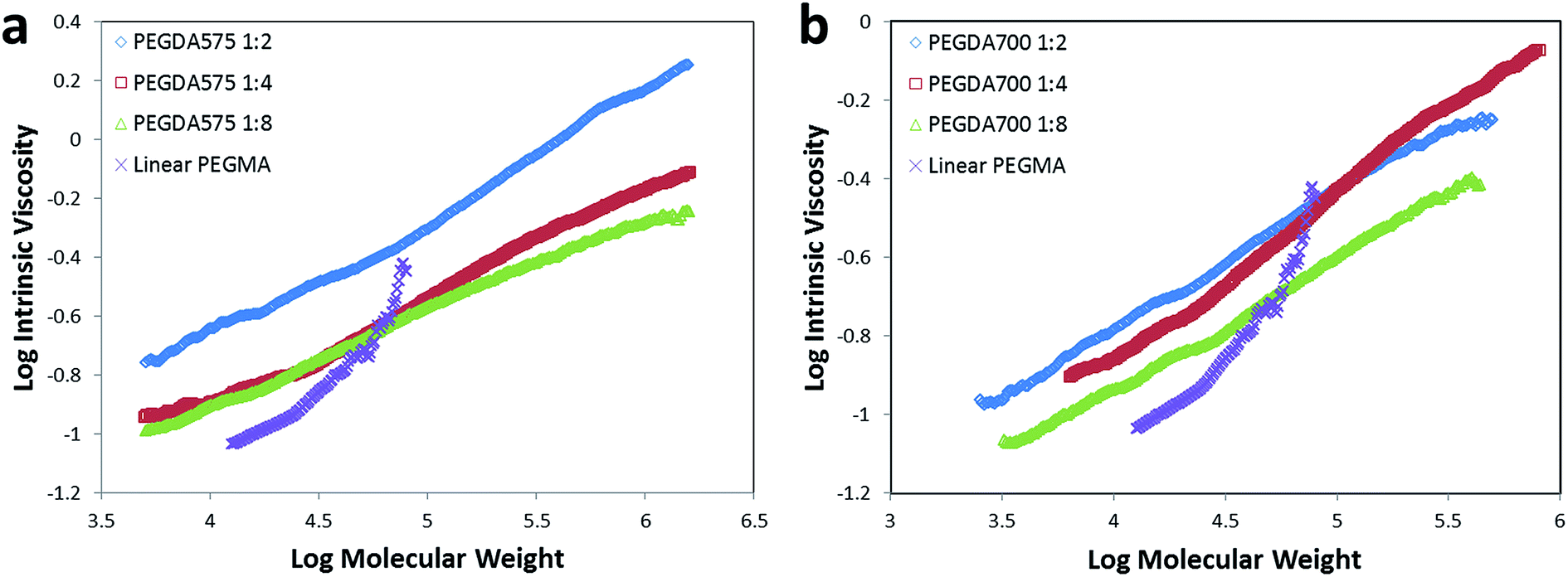 | ||
| Fig. 2 Mark–Houwink plots for the polymers obtained by homopolymerization of (a) PEGDA575 and (b) PEGDA700 using different initiator-to-monomer ratios. The Mark–Houwink plots of a linear polymer from ATRP of poly(ethylene glycol) methyl ether acrylate (PEGMA, Mn = 480) is presented in the figures for comparison. | ||
1H-NMR analysis (Fig. 3 and eqn (S1)–(S3) in ESI†) for the poly(PEGDA)s demonstrates the existence of a high amount of vinyl functional groups at characteristic peaks between 6.5 ppm and 5.7 ppm. The vinyl content and branch ratio is outlined in Table 1. The vinyl content decreases reasonably with increasing the initiator-to-monomer ratio because more vinyl groups are consumed by the addition to the halogen-containing initiator at the early stage and by the chain combination at the later stage. The calculations also showed that these polymers possess a high degree of branching, with the highest value of 62.7% for poly(PEGDA575) 1:2 (Table 1). This value indicates that ∼6 branching unit exists for every 10 PEGDA units linked together in a –C–C– chain. The initiator contents of the polymers were also summarized in Table 1. The ratios of the initiator/PEGDA units were generally proportional to their initial feed ratios. The polymers with initial feed ratios of 1:4 and 1:8 contain higher initiator contents than the theoretical contents due to the high initiation efficiency and the incomplete conversion of PEGDA monomers. In contrast, the polymers with initial feed ratios of 1:2 contain lower initiator contents than the theoretical contents possibly because of the consumption of initiator from the termination reaction at the early stage due to the high initiator concentration. The initiator content in the polymer products is lower than the content of the branching unit for all the poly(PEGDA)s, indicating that more than one connection per primary chain exists and that there is still a certain number of ‘loops’ existing in the polymer products. The formation of the ‘loops’ could be attributed to the secondary intramolecular reaction at later stage when the local concentration of both pendent vinyl and initiation site increased. The flexibility of the PEGDA units and the increasing mobility of macromolecules at later stage also might account for the intramolecular reaction. The proportion of the ‘loops’ in the polymer can be calculated as the proportion of the branching units over the initiator units. It is noteworthy that the proportions of ‘loops’ in poly(PEGDA575)s (∼20%) are higher than that in poly(PEGDA700)s (∼15%), giving the speculation that the diacrylate with shorter lengths of PEG spacers may induce more intramolecular reaction due to higher local concentration of pendent vinyl groups. Free vinyl groups were also left within the poly(PEGDA)s (Fig. 3), which can be used for post functionalization via thiol–ene click chemistry approach.
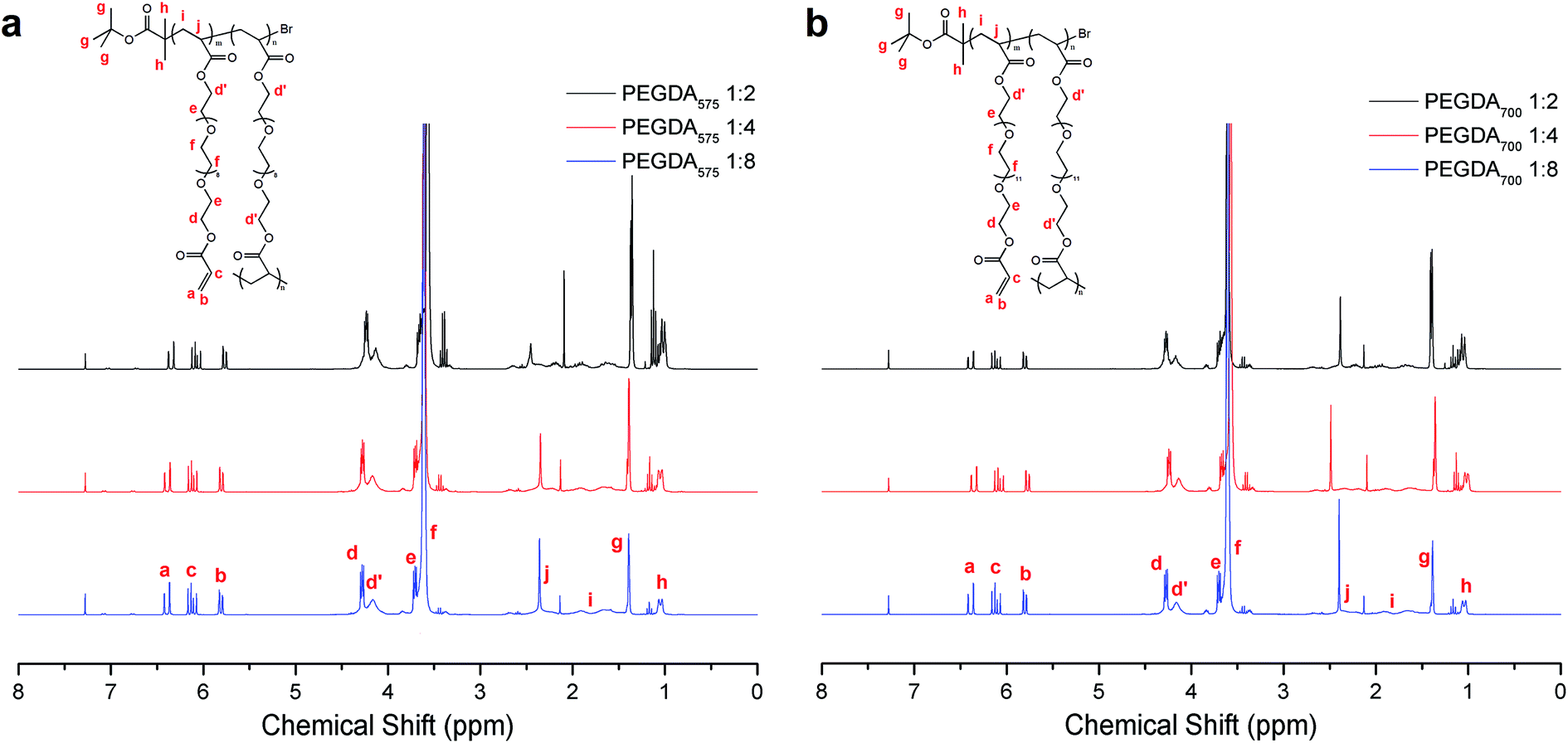 | ||
| Fig. 3 1H NMR spectroscopy of purified polymers obtained by homopolymerization of (a) PEGDA575 and (b) PEGDA700 with different initiator-to-monomer ratios. | ||
Both poly(PEGDA575)s and poly(PEGDA700)s were soluble in polar solvents (including water and methanol) as well as in many organic solvents (e.g. THF and chloroform). The products obtained from PEGDA700 show good solubility in aqueous solution due to the longer PEG chains which can provide a higher hydrophilicity to the molecules, whereas the products from PEGDA575 were found to exhibit a characteristic thermoresponsive property in distilled water. When the temperature was raised to a certain value, the polymers from PEGDA575 were precipitated out from water and settled into another layer after several minutes. The polymers were also able to reversely dissolve when the temperature dropped back. The thermally induced phase separation behavior in water was monitored by raising the temperature from 15 to 80 °C and measuring the temperature at the onset of cloudiness. It is well known that thermoresponsive polymer chains in solution adapt an expanded coil conformation and they collapse at phase transition temperature to form compact globules. The globules aggregate in the absence of mechanisms that reduce surface tension, subsequently causing turbidity and the formation of visible particles.37 Many previous studies have explored the phase transition temperature for PEG based polymers in a low polymer concentration (typically 0.2% w/v). However, we found that the phase transition temperature of the poly(PEGDA575)s is also strongly dependent on polymer concentration.37
As shown in Fig. 4(a), the lower critical solution temperatures (LCSTs) for the poly(PEGDA575)s with different initiator/monomer ratio (1:2, 1:4 and 1:8) appear at the polymer concentration of ∼2.5% w/v. The LCST values are 9 °C, 21 °C and 31 °C for poly(PEGDA575) 1:2, poly(PEGDA575) 1:4 and poly(PEGDA575) 1:8, respectively. The initiator end group and the carbon–carbon backbone are hydrophobic components, whereas the PEG chains are hydrophilic components. We believe that the difference in LCST is mainly attributed to the different hydrophobic/hydrophilic composition of the products, since more PEGDA units would enhance the polymer–water hydrogen bonding interaction and thus expand the temperature range of miscibility, whereas more initiator end group or longer backbone would lower the LCSTs by bringing higher thermodynamic cost of solvation. It is worth mentioning that as the backbone length increases, the number of PEG chains will also increase and thus the influence of the backbone is less significant than that of the initiator which makes the main hydrophobic contribution on different polymers.
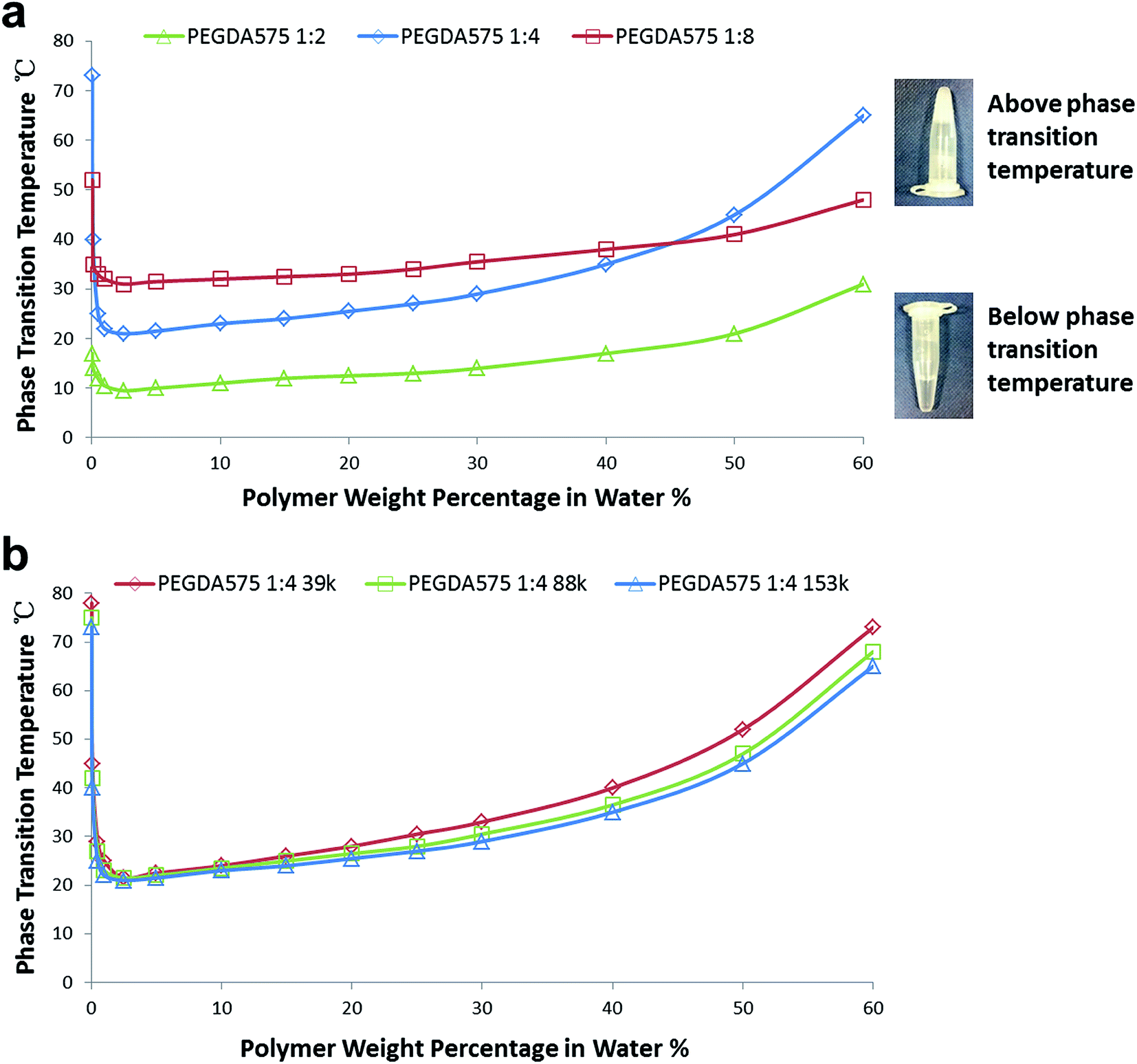 | ||
| Fig. 4 Thermoresponsive properties of the obtained poly(PEGDA575)s: (a) phase transition temperature of the poly(PEGDA575)s in distilled water at different concentrations. (b) Comparison of phase transition temperature of the poly(PEGDA575)s 1:4 obtained at different times of polymerization with different molecular weight. | ||
It can be noted that the phase transition temperature for all of the three poly(PEGDA575)s changed with a similar trend, but with different changing rates. The poly(PEGDA575) 1:4 increased dramatically from 21 °C to 65 °C with the polymer concentration raised from 2.5% to 60% w/v. However, the increase is more gentle for the poly(PEGDA575) 1:2 and the poly(PEGDA575) 1:8. It is known that the phase transition temperature of a polymer is dependent upon a series of factors affecting solubility. And it has been reported that increasing molecular weight (Mw) tends to depress the phase transition temperature and broadens the phase transition lines due to an increasing energy cost of solvation.38–40 This could explain why the poly(PEGDA575) 1:2 which has a Mw of 403 kDa shows a more steady line than the poly(PEGDA575) 1:4 which has a Mw of 153 kDa. This tendency is well confirmed in Fig. 4(b), in which the phase transition behavior of poly(PEGDA575) 1:4 with different Mw were studied. The polymers were prepared at different time points of polymerization and summarized in Table S2.† The branch ratios for these polymers were similar but the molecular weight showed a significant difference. As can be seen in Fig. 4(b), with an increase in Mw, the phase transition temperature drops slightly at each polymer concentration and the phase transition line becomes broader.
The poly(PEGDA575) 1:8 also showed a steady change of phase transition temperature despite of having the lowest Mw. This could be attributed to the longer hydrophobic backbone in the poly(PEGDA575) 1:8. It can be noted that the phase transition temperature for the poly(PEGDA575) 1:8 is maintained between 30 °C to 40 °C for a wide range of concentration. This phase transition property around body temperature holds great potential for biomedical applications in various areas such as smart hydrogels and drug delivery.
Conclusion
A series of water soluble hyperbranched polymers were prepared by homopolymerization of PEGDA through enhanced intermolecular branching. High monomer conversions (up to 96%) and hyperbranched polymer structure were both achieved by increasing the ratio of initiator/monomer, which results in a high chain concentration and a smaller chain dimension before the crosslinking reaction occurs. The poly(PEGDA700)s show good solubility in aqueous solution whereas the poly(PEGDA575)s show a characteristic thermoresponsive property, which is highly dependent on the polymer concentration and the polymer composition. These water-soluble thermoresponsive vinyl functional polymers have the potential for further modification towards applications in drug delivery or biosensors. The polymerization strategy described here could be broadly applicable to a large number of polymerization techniques and different MVMs, favoring the production of veritable hyperbranched polymers in one pot reactions.Acknowledgements
Health Research Board (HRB) of Ireland (HRA-POR-2013-412; HRA/2009/121/R) and Science Foundation Ireland (SFI), SFI Principal Investigator programme (10/IN.1/B2981(T)), DEBRA Ireland, University College Dublin, Strategic and Major Initiative 2014 (Scholarship), are gratefully acknowledged for funding.References
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef.
- R. Baudry and D. C. Sherrington, Macromolecules, 2006, 39, 1455–1460 CrossRef CAS.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, J. Mater. Chem., 2003, 13, 2701–2710 RSC.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, Macromolecules, 2004, 37, 2096–2105 CrossRef CAS.
- S. Camerlynck, P. A. G. Cormack, D. C. Sherrington and G. Saunders, J. Macromol. Sci., Part B: Phys., 2005, 44, 881–895 CrossRef CAS PubMed.
- P. A. Costello, I. K. Martin, A. T. Slark, D. C. Sherrington and A. Titterton, Polymer, 2002, 43, 245–254 CrossRef CAS.
- J. Rosselgong and S. P. Armes, Macromolecules, 2012, 45, 2731–2737 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43, 2145–2156 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. Barton and D. Price, Macromolecules, 2009, 42, 5919–5924 CrossRef CAS.
- Y. Li, A. J. Ryan and S. P. Armes, Macromolecules, 2008, 41, 5577–5581 CrossRef CAS.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483–7492 CrossRef CAS.
- Y. T. Li and S. P. Armes, Macromolecules, 2005, 38, 8155–8162 CrossRef CAS.
- Y. T. Li and S. P. Armes, Macromolecules, 2005, 38, 5002–5009 CrossRef CAS.
- L. Wang, C. M. Li, A. J. Ryan and S. P. Armes, Adv. Mater., 2006, 18, 1566–1570 CrossRef CAS PubMed.
- H. Gao, P. Polanowski and K. Matyjaszewski, Macromolecules, 2009, 42, 5925–5932 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS PubMed.
- H. Gao, W. Li and K. Matyjaszewski, Macromolecules, 2008, 41, 2335–2340 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087–3092 CrossRef CAS.
- W. Li, J. A. Yoon, M. Zhong and K. Matyjaszewski, Macromolecules, 2011, 44, 3270–3275 CrossRef CAS.
- M. Akiyama, K. Yoshida and H. Mori, Polymer, 2014, 55, 813–823 CrossRef CAS PubMed.
- Y. Lin, X. Liu, X. Li, J. Zhan and Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 26–40 CrossRef CAS PubMed.
- W. Wang, Y. Zheng, E. Roberts, C. J. Duxbury, L. Ding, D. J. Irvine and S. M. Howdle, Macromolecules, 2007, 40, 7184–7194 CrossRef CAS.
- T. Zhao, Y. Zheng, J. Poly and W. Wang, Nat. Commun., 2013, 4, 1873 CrossRef PubMed.
- Y. Zheng, H. Cao, B. Newland, Y. Dong, A. Pandit and W. Wang, J. Am. Chem. Soc., 2011, 133, 13130–13137 CrossRef CAS PubMed.
- Y. Zheng, B. Newland, H. Tai, A. Pandit and W. Wang, Chem. Commun., 2012, 48, 3085–3087 RSC.
- H. Mori and M. Tsukamoto, Polymer, 2011, 52, 635–645 CrossRef CAS PubMed.
- Q. Yu, F. Q. Zeng and S. P. Zhu, Macromolecules, 2001, 34, 1612–1618 CrossRef CAS.
- M. L. Koh, D. Konkolewicz and S. Perrier, Macromolecules, 2011, 44, 2715–2724 CrossRef CAS.
- D. T. Landin and C. W. Macosko, Macromolecules, 1988, 21, 846–851 CrossRef CAS.
- R. Wang, Y. Luo, B.-G. Li and S. Zhu, Macromolecules, 2009, 42, 85–94 CrossRef CAS.
- R. Saito, Y. Iijima and K. Yokoi, Macromolecules, 2006, 39, 6838–6844 CrossRef CAS.
- R. Saito, Polymer, 2008, 49, 2625–2631 CrossRef CAS PubMed.
- Z.-M. Dong, X.-H. Liu, Y. Lin and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6023–6034 CrossRef CAS PubMed.
- Z.-M. Dong, X.-H. Liu, X.-L. Tang and Y.-S. Li, Macromolecules, 2009, 42, 4596–4603 CrossRef CAS.
- Y. Bao, G. Shen, X. Liu and Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2959–2969 CrossRef CAS PubMed.
- I. Bannister, N. C. Billingham and S. P. Armes, Soft Matter, 2009, 5, 3495–3504 RSC.
- S. Z. D. Cheng, Phase Transitions in Polymers: The Role of Metastable States, 2008 Search PubMed.
- Y. C. Bae, S. M. Lambert, D. S. Soane and J. M. Prausnitz, Macromolecules, 1991, 24, 4403–4407 CrossRef CAS.
- F. E. Bailey and R. W. Callard, J. Appl. Polym. Sci., 1959, 1, 56–62 CrossRef CAS PubMed.
- D. R. Yen, S. Raghavan and E. W. Merrill, Macromolecules, 1996, 29, 8977–8978 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01253h |
| This journal is © The Royal Society of Chemistry 2015 |