
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed silylation of p-quinone methides: new entry to dibenzylic silanes†
Aurora
López
,
Alejandro
Parra
,
Carlos
Jarava-Barrera
and
Mariola
Tortosa
*
Departamento de Química Orgánica, Universidad Autónoma de Madrid Cantoblanco, 28049 Madrid, Spain. E-mail: mariola.tortosa@uam.es
First published on 22nd October 2015
Abstract
An efficient and general copper(I)-catalyzed silylation of p-quinone-methides is described. Non-symmetric dibenzylic silanes are obtained in high yields under mild reaction conditions. These compounds can be used as bench-stable benzylic carbanion precursors.
para-Quinone methides are reactive intermediates formed by a cyclohexadiene moiety in para-conjugation with a carbonyl group and an exo-methylene component. They are neutral entities with a zwitterionic resonance structure that enhances the electrophilic character at the δ-position.1 Carbanions, aromatic rings, alcohols and amines are typical nucleophiles that quickly react with p-quinone methides to afford a variety of diaryl derivatives (Scheme 1).2 The use of transition metals could allow the formation of C–C and C–X bonds complementary to those formed by direct addition of a typical nucleophile. Surprisingly, the use of metal-catalyzed transformations to functionalize the exocyclic double bond of p-quinone methides remains largely unexplored.3
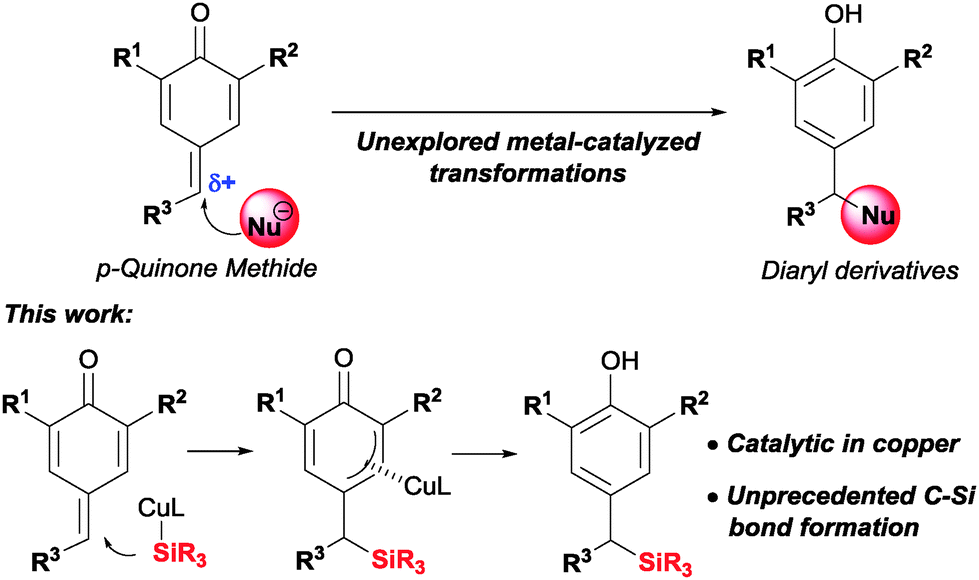 | ||
| Scheme 1 p-Quinone methides as diaryl derivatives precursors. | ||
We became interested in probing this approach using a silyl copper(I) complex as a formal silicon nucleophile (Scheme 1). To the best of our knowledge, the addition of nucleophilic silicon species to ortho- or para-quinone methides has not been studied to date. Silicon-containing molecules are valuable synthetic intermediates which can be converted into useful compounds through a number of transformations.4 Recently, copper-catalyzed silylation reactions have emerged as a powerful tool for C–Si bond formation.5 We envisioned that insertion of the exocyclic double bond into the Cu–Si bond followed by aromatization would afford non-symmetric benzylic silanes.
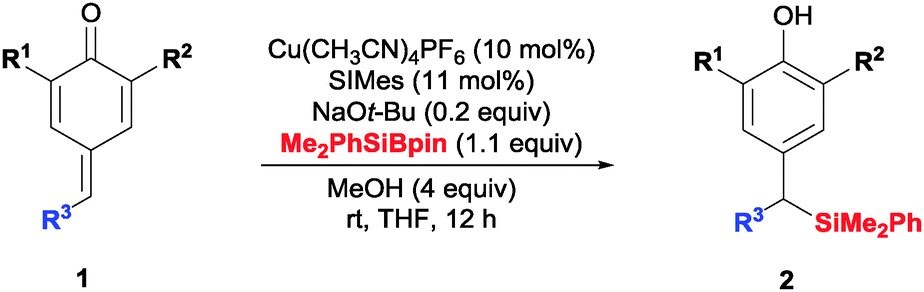
The most common way to synthesize benzylic silanes involves the reaction between an in situ generated benzylic carbanion and a silyl chloride.6 Our method would offer a milder alternative to this classic approach, avoiding the use of stoichiometric amounts of strong bases. Herein, we describe a copper(I)-catalyzed protocol for the silylation–aromatization of p-quinone-methides. The reactions proceed in high yields using only 10% of an inexpensive copper(I) salt and a commercially available silaborane reagent.7
We started our study with p-quinone methide 1a, containing removable t-Bu groups at the α-positions (Table 1).8 A series of ligands were screened (Table 1, entries 1–6) using Cu(CH3CN)4PF6 (10 mol%), Me2PhSiBpin (1.1 equiv.), NaOt-Bu (0.2 equiv.) and MeOH (4 equiv.). We found that NHC ligands (entries 5 and 6) were superior to monodentate or bidentate phosphines (entries 1–4). SIMes gave the best results, affording dibenzylic silane 2a with 86% isolated yield (entry 6, ≥98% conversion). The use of other bases (entries 7–9) or different copper salts (entries 10 and 11) gave poorer results. Lowering the catalyst loading to 5 mol% also resulted in a significantly lowered yield (entry 12). In the absence of MeOH (entry 13) or with only two equivalents (entry 14) compound 2a was obtained in 46% and 57% yield respectively. Finally, to check the role of the NHC–Cu(I) catalyst we carried out the reaction in the absence of copper salt and ligand (entry 15). Under those conditions, a very complex mixture was observed in the 1H NMR spectrum of the crude product. From this mixture, we could identify the product of 1,6-addition of methoxide to 1a as the main compound, unreacted 1a, and a small amount of 2a. The formation of 2a under these conditions could be explained by alkoxide activation of the silaborane in the absence of the copper catalyst.9
|
|
||||
|---|---|---|---|---|
| Entrya | Copper salt | Base | Ligand | 2a (%) |
| a Reaction conditions: 1a (0.2 mmol), Me2PhSiBpin (0.22 mmol), base (20 mol%), Cu(CH3CN)4PF6 (10 mol%), ligand (11 mol%), MeOH (0.8 mmol), THF (0.1 M). b Conversion determined by 1H NMR analysis of the crude mixture. c Yield of isolated 2a. d Reaction conditions: 1a (0.2 mmol), Me2PhSiBpin (0.22 mmol), NaOt-Bu (20 mol%), Cu(CH3CN)4PF6 (5 mol%), ligand (11 mol%), MeOH (0.8 mmol), THF (0.1 M). e The reaction was carried out in the absence of MeOH. f 0.4 mmol of MeOH were used. g Reaction conditions: 1a (0.2 mmol), Me2PhSiBpin (0.22 mmol), NaOt-Bu (20 mol%), MeOH (0.8 mmol), THF (0.1 M). | ||||
| 1 | Cu(CH3CN)4PF6 | NaOt-Bu | Ph3P | 64b |
| 2 | Cu(CH3CN)4PF6 | NaOt-Bu | JohnPhos | 64b |
| 3 | Cu(CH3CN)4PF6 | NaOt-Bu | Xantphos | 50b |
| 4 | Cu(CH3CN)4PF6 | NaOt-Bu | (±)-BINAP | 45b |
| 5 | Cu(CH3CN)4PF6 | NaOt-Bu | IMes | 87b |
| 6 | Cu(CH 3 CN) 4 PF 6 | NaOt-Bu | SIMes | ≥98 (86) |
| 7 | Cu(CH3CN)4PF6 | KOt-Bu | SIMes | 60c |
| 8 | Cu(CH3CN)4PF6 | LiOt-Bu | SIMes | 68c |
| 9 | Cu(CH3CN)4PF6 | CsF | SIMes | 61c |
| 10 | CuCl | NaOt-Bu | SIMes | 55c |
| 11 | Cu2O | NaOt-Bu | SIMes | 20c |
| 12d | Cu(CH3CN)4PF6 | NaOt-Bu | SIMes | 35c |
| 13e | Cu(CH3CN)4PF6 | NaOt-Bu | SIMes | 46c |
| 14f | Cu(CH3CN)4PF6 | NaOt-Bu | SIMes | 57c |
| 15g | — | NaOt-Bu | — | — |

With these optimal conditions in hand, we proceeded to study the scope of the silylation–aromatization process (Table 2). We first modified the stereoelectronic properties of the exomethylene substituent (R3). Dibenzylic silanes with electron donating groups (compounds 2b–2c), heterocycles (compound 2d), and a larger naphthyl group (compound 2e) were prepared in high yields. The conditions also worked for p-quinone methides with electron withdrawing groups in para (compounds 2f, 2i, 2j), ortho (compound 2g) and meta (compound 2h) positions. It should be pointed out that our method allows for the synthesis of compounds with halogen substituents (2f, 2g) and an ester group (2i), which would be difficult to obtain by the reaction of a dibenzylic carbanion and a silyl chloride. Interestingly, monobenzylic silane 2k, in which R3 is an alkyl group, was also obtained using the optimized conditions.

Additionally, we modified the R1 and R2 substituents. Compounds 2l and 2m, with two methyl groups, and compound 2n, with two isopropyl groups, were obtained in good yields. It is also possible to introduce two different alkyl groups in the α-position (compound 2o) starting from a non-symmetrical p-quinone methide. Finally, the structure of compound 2g was confirmed by single crystal X-ray crystallography (Fig. 1).
 | ||
| Fig. 1 X-ray structure of compound 2g. | ||
One interesting feature of benzylic silanes is their ability to be used as bench-stable benzylic anion equivalents under mild reaction conditions.10 However, most known examples of these transformations have been performed with monobenzylic trimethylsilane derivatives. Therefore, our method provided an opportunity to check if dibenzylic dimethylphenyl silanes such as 2 could be also used as carbanion precursors. To the best of our knowledge, the generation of dibenzylic carbanions from silanes has not previously been reported. Gratifyingly, treatment of silane 2a with cesium fluoride in DMF, followed by addition of p-chloro benzaldehyde, provided the desired compound 3 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers (Scheme 2). Oxidation followed by removal of the t-butyl groups8 using AlCl3 afforded α,α-diaryl ketone 4 in a good overall yield.
:1 mixture of diastereomers (Scheme 2). Oxidation followed by removal of the t-butyl groups8 using AlCl3 afforded α,α-diaryl ketone 4 in a good overall yield.
 | ||
| Scheme 2 Functionalization of the C–Si bond and de-tert-butylation. | ||
A possible mechanism for the silylation–aromatization reaction of p-quinone methides is shown in Scheme 3. First, a silyl-Cu(I)–NHC complex B is formed by reaction of a copper alkoxide A and the silaborane reagent. Insertion of the exocyclic double bond of the p-quinone methide into the Cu–Si bond affords a π-allyl-copper intermediate (C) that could isomerize to copper phenoxide E. At this point two pathways are possible. Protonolysis in the presence of MeOH would provide silane 2 with release of NHC–CuOMe to restart the catalytic cycle. On the other hand, copper phenoxide E could react directly with the silaborane to provide 2 and silyl-copper complex B.
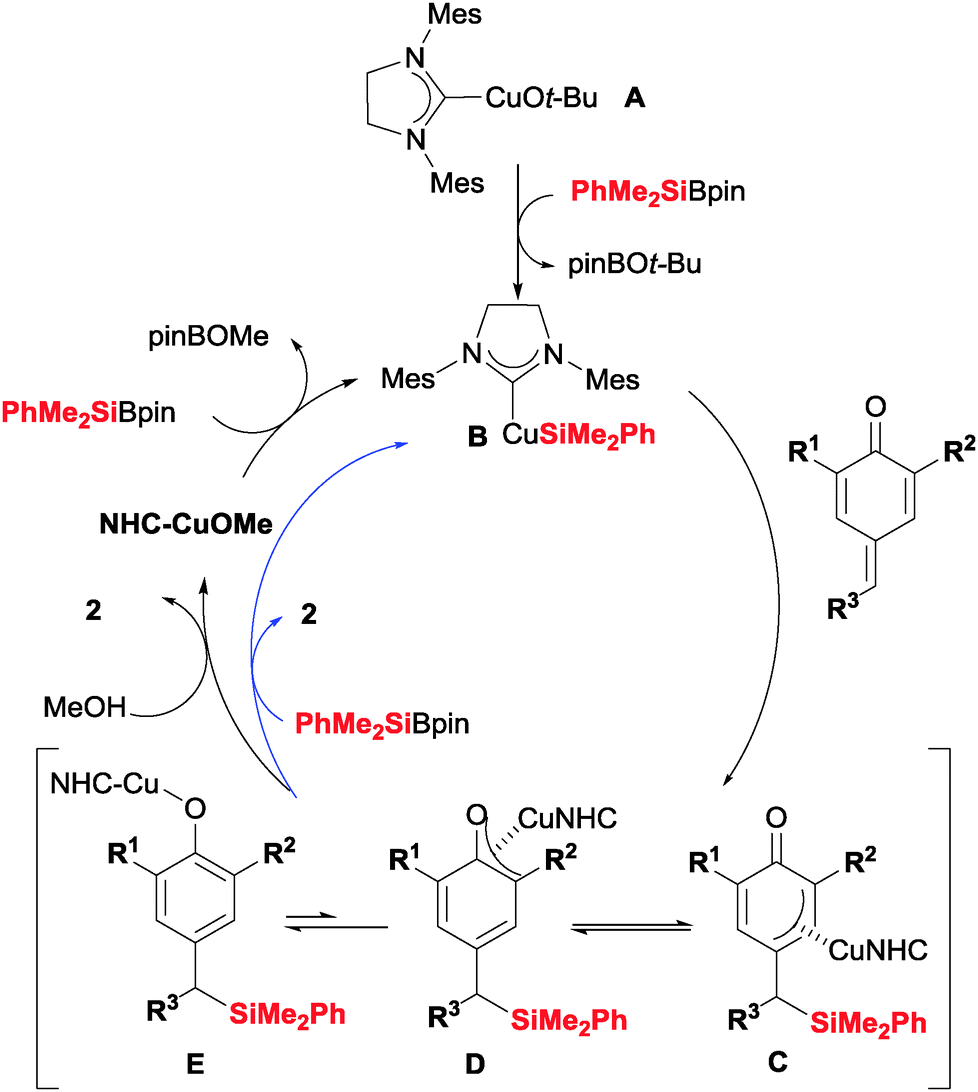 | ||
| Scheme 3 Plausible mechanism for the silylation–aromatization. | ||
In conclusion, we have found that copper(I) salts can catalyze the silylation–aromatization process of p-quinone methides. This study represents the first silicon addition to a quinone methide and provides new insight for the development of novel metal-catalyzed transformations. Mono- and dibenzylic silanes can be prepared in high yields under mild reaction conditions. We have also demonstrated that dibenzylic silanes can be used as stable dibenzylic carbanion equivalents. The development of asymmetric versions of this and related transformations is underway.
We thank the European Research Council (ERC-337776) and MINECO (CTQ2012-35957) for financial support. M. T. and A. P. thank MICINN for RyC and JdC contracts. We acknowledge Dr Josefina Perles for X-ray structure analysis.
Notes and references
- For reviews and highlights of p-quinone methides: (a) H.-U. Wagner and R. Gompper, in The Chemistry of the Quinonoid Compounds, ed. S. Patai, Wiley, New York, 1974, ch. 18, vol. 2, p. 1145 Search PubMed; (b) M. M. Toteva and J. P. Richard, Adv. Phys. Org. Chem., 2011, 45, 39 CrossRef CAS PubMed; (c) A. Parra and M. Tortosa, ChemCatChem, 2015, 7, 1524 CrossRef CAS PubMed; (d) L. Caruana, M. Fochi and L. Bernardi, Molecules, 2015, 20, 11733 CrossRef CAS PubMed.
- For selected examples: (a) L. J. Filar and S. Winstein, Tetrahedron Lett., 1960, 25, 9 Search PubMed; (b) D. J. Hart, P. A. Cain and D. A. Evans, J. Am. Chem. Soc., 1978, 100, 1548 CrossRef CAS; (c) S. R. Angle and K. D. Turnbull, J. Am. Chem. Soc., 1989, 111, 1136 CrossRef CAS; (d) R. Lucius, R. Loos and H. Mayr, Angew. Chem., Int. Ed., 2002, 41, 92 CrossRef; (e) T. Bug and H. Mayr, J. Am. Chem. Soc., 2003, 125, 12980 CrossRef CAS PubMed; (f) S. T. A. Berger, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2007, 129, 9753 CrossRef CAS PubMed; (g) D. Richter, N. Hampel, T. Singer, A. R. Ofial and H. Mayr, Eur. J. Org. Chem., 2009, 3203 CrossRef CAS PubMed; (h) R. Appel, R. Loos and H. Mayr, J. Am. Chem. Soc., 2009, 131, 704 CrossRef CAS PubMed; (i) T. A. Nigst, A. Antipova and H. Mayr, J. Org. Chem., 2012, 77, 8142 CrossRef CAS PubMed; (j) D. S. Allgäuer, P. Mayer and H. Mayr, J. Am. Chem. Soc., 2013, 135, 15216 CrossRef PubMed; (k) W.-D. Chu, L.-F. Zhang, X. Bao, X.-H. Zhao, C. Zeng, J.-Y. Du, G.- B. Zhang, F.-X. Wang, X.-Y. Ma and C.-A. Fan, Angew. Chem., Int. Ed., 2013, 52, 9229 CrossRef CAS PubMed; (l) L. Caruana, F. Kniep, T. K. Johasen, P. H. Poulsen and K. A. Jørgensen, J. Am. Chem. Soc., 2014, 136, 15929 CrossRef CAS PubMed; (m) Z. Wang, F. Ai, Z. Wang, W. Zhao, G. Zhu, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 383 CrossRef CAS PubMed.
- For the stabilization of p-quinone methides by complexation with transition-metals, see: (a) A. Vigalok, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 1998, 120, 477 CrossRef CAS; (b) O. Rabin, A. Vigalok and D. Milstein, Chem. – Eur. J., 2000, 6, 454 CrossRef CAS.
- (a) E. W. Colvin, Silicon Reagents in Organic Synthesis, Academic Press, London, 1988 Search PubMed; (b) Chemistry of Organosilicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley-VCH, New York, vol. 3, 2001 Search PubMed; (c) W. Bains and R. Tacke, Curr. Opin. Drug Discovery Dev., 2003, 6, 526 CAS; (d) G. A. Showwell and J. S. Mills, Drug Discovery Today, 2003, 8, 551 CrossRef; (e) M. Mortensen, R. Husmann, E. Veri and C. Bolm, Chem. Soc. Rev., 2009, 38, 1002 RSC.
- For recent copper-catalyzed reactions using a silaborane reagent: (a) K.-S. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898 CrossRef CAS PubMed; (b) D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 8513 CrossRef CAS PubMed; (c) A. Welle, J. Petrignet, B. Tinant, J. Wouters and O. Riant, Chem. – Eur. J., 2010, 16, 10980 CrossRef CAS PubMed; (d) C. Kleeberg, E. Feldmann, E. Hartmann, D. J. Vyas and M. Oestreich, Chem. – Eur. J., 2011, 17, 13538 CrossRef CAS PubMed; (e) D. J. Vyas, C. K. Hazra and M. Oestreich, Org. Lett., 2011, 13, 4462 CrossRef CAS PubMed; (f) D. J. Vyas, R. Fröhlich and M. Oestreich, Org. Lett., 2011, 13, 2094 CrossRef CAS PubMed; (g) C. Kleeberg, M. S. Cheung, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 19060 CrossRef CAS PubMed; (h) K.-S. Lee, H. Wu, F. Haeffner and A. H. Hoveyda, Organometallics, 2012, 31, 7823 CrossRef CAS PubMed; (i) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487 CrossRef CAS PubMed; (j) F. Meng, H. Jang and A. H. Hoveyda, Chem. – Eur. J., 2013, 19, 3204 CrossRef CAS PubMed; (k) C. K. Hazra, E. Irran and M. Oestreich, Eur. J. Org. Chem., 2013, 4903 CrossRef CAS PubMed; (l) L. B. Delvos, D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 4650 CrossRef CAS PubMed; (m) V. Pace, J. P. Rae, H. Y. Harb and D. J. Procter, Chem. Commun., 2013, 49, 5150 RSC; (n) V. Pace, J. P. Rae and D. J. Procter, Org. Lett., 2013, 15, 476 CrossRef PubMed; (o) V. Cirriez, C. Rasson, T. Hermant, J. Petrignet, J. Díaz Álvarez, K. Robeyns and O. Riant, Angew. Chem., Int. Ed., 2013, 52, 1785 CrossRef CAS PubMed; (p) S. Vercruysse, L. Cornelissen, F. Nahra, L. Collard and O. Riant, Chem. – Eur. J., 2014, 20, 1834 CrossRef CAS PubMed; (q) Y.-H. Xu, L.-H. Wu, J. Wang and T.-P. Loh, Chem. Commun., 2014, 50, 7195 RSC; (r) A. Hensel, K. Nagura, L. B. Delvos and M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 4964 CrossRef CAS PubMed; (s) J. Rae, Y. C. Hu and D. J. Procter, Chem. – Eur. J., 2014, 20, 13143 CrossRef CAS PubMed; (t) A. García-Rubia, J. A. Romero-Revilla, P. Mauleón, R. Gómez-Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2015, 137, 6857 CrossRef PubMed.
- (a) A. R. Katritzky and M. Qi, J. Org. Chem., 1997, 62, 4116 CrossRef CAS; (b) T. R. van den Ancker, C. L. Raston, B. W. Skelton and A. H. White, Organometallics, 2000, 19, 4437 CrossRef CAS; (c) V. Georgakilas, G. P. Perdikomatis, A. S. Triantafyllon, M. G. Siskos and A. K. Zarkadis, Tetrahedron, 2002, 58, 2441 CrossRef CAS; (d) M. S. Hill and P. B. Hitchcock, Organometallics, 2002, 21, 220 CrossRef CAS; (e) N. S. Mills, C. Tirla, M. A. Benish, A. J. Rakowitz, L. M. Bebell, C. M. M. Hurd and A. L. M. Bria, J. Org. Chem., 2005, 70, 10709 CrossRef CAS PubMed; (f) J. A. Wilkinson, S. B. Rossington, S. Ducki, J. Leonardb and N. l. Hussain, Tetrahedron, 2006, 62, 1833 CrossRef CAS PubMed; (g) Y. S. Park, E. K. Yum, A. Basu and P. Beak, Org. Lett., 2006, 8, 2667 CrossRef CAS PubMed ; for an exception using iridium-catalyzed C–H activation: ; (h) Q. Li, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2014, 53, 8471 CrossRef CAS PubMed ; for an exception using Ni/Cu-catalyzed C–O cleavage: ; (i) C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236 CrossRef CAS PubMed.
- (a) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29 CrossRef CAS; (b) M. Suginome and Y. J. Ito, Organomet. Chem., 2003, 680, 43 CrossRef CAS . For a review on the chemistry of silaboranes, see: ; (c) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402 CrossRef CAS PubMed.
- For a review on the use of the t-Bu group as a protecting group in the synthesis of aromatic compounds, see: S. A. Saleh and H. I. Tashtoush, Tetrahedron, 1998, 54, 14157 CrossRef CAS.
- (a) J. M. O’Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712 CrossRef PubMed; (b) H. Ito, Y. Horita and E. Yamamoto, Chem. Commun., 2012, 48, 8006 RSC; (c) K. Oshima, T. Ohmura and M. Suginome, Chem. Commun., 2012, 48, 8571 RSC; (d) C. Kleeberg and C. Borner, Eur. J. Inorg. Chem., 2013, 2799 CrossRef CAS PubMed.
- (a) A. Ricci, A. Degl’innocenti, M. Fiorenza, M. Taddei and M. A. Spartera, Tetrahedron Lett., 1982, 23, 577 CrossRef CAS; (b) B. Bennetau and J. Dunogues, Tetrahedron Lett., 1983, 24, 4217 CrossRef CAS; (c) G. Bartoli, M. Bosco, D. Caretti, R. Dalpozzo and P. E. Todesco, J. Org. Chem., 1987, 52, 4381 CrossRef CAS; (d) A. S. Pilcher and P. DeShong, J. Org. Chem., 1996, 61, 6901 CrossRef CAS PubMed; (e) S. Thayumanavan, Y. S. Park, P. Farid and P. Beak, Tetrahedron Lett., 1997, 38, 5429 CrossRef CAS; (f) M. Das and D. F. O’Shea, Tetrahedron, 2013, 69, 6448 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and crystallographic data. CCDC 1414585. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc06653k |
| This journal is © The Royal Society of Chemistry 2015 |