
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanism of a one-photon two-electron process in photocatalytic hydrogen evolution from ascorbic acid with a cobalt chlorin complex†
Shoko
Aoi
a,
Kentaro
Mase
a,
Kei
Ohkubo
ab and
Shunichi
Fukuzumi
*abc
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370
bDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea
cFaculty of Science and Technology, Meijo University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Nagoya, Aichi 468-8502, Japan
First published on 21st August 2015
Abstract
A one-photon two-electron process was made possible in photocatalytic H2 evolution from ascorbic acid with a cobalt(II) chlorin complex [CoII(Ch)] via electron transfer from ascorbate to the excited state of [Ru(bpy)3]2+ followed by electron transfer from [Ru(bpy)3]+ to CoII(Ch) with proton to give the hydride complex, which reacts with proton to produce H2. [CoIII(Ch)]+ was reduced by ascorbate to reproduce CoII(Ch).
Photocatalytic production of hydrogen (H2) has attracted increasing attention as a clean energy source because of the ever-increasing demand for energy and climate change on our planet.1 A number of highly efficient hydrogen evolving systems have been developed including homogeneous and heterogeneous photocatalytic systems.2–13 Two electrons are required to produce H2 from protons, although one photon generates normally only one electron. A mechanism of photocatalytic production of H2 was reported to clarify how photoinduced electron transfer of a photosensitiser (a one-electron process) leads to H2 production (a two-electron process).14–16 Disproportionation of one-electron reduced species of metal complexes resulted in formation of the two-electron reduced species from which H2 is formed.17 Bimolecular reactions of metal(III)–hydride complexes also generate H2 accompanied by regeneration of metal(II) complexes.18 In each case, the maximum quantum yield of H2 production per photon is 50%, because two photons are required to produce two electrons. Thus there has so far been no example for one photon to generate one H2 molecule.
We report herein photocatalytic H2 evolution from ascorbic acid (AscH2) with a cobalt(II) chlorin complex [CoII(Ch)] (a chemical structure shown in Scheme 1)19 in an aqueous acetonitrile solution (H2O/MeCN), which proceeds via a one-photon two-electron process. The photocatalytic mechanism is clarified by nanosecond laser transient absorption spectra and by examining each step in the catalytic cycle independently.
Visible light irradiation of a deaerated (Ar-saturated) H2O/MeCN solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) of [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) containing ascorbic acid (AscH2) and ascorbate (AscH−) (Eox = 0.43 V vs. SCE) as an electron donor and CoII(Ch) (Ered = −0.96 V vs. SCE) (Fig. S1 in the ESI†) as a catalyst resulted in H2 evolution (Fig. 1, black line). When the ratio of AscH− to AscH2 was changed as fixed total concentrations of AscH2 and AscH− ([AscH2] + [AscH−] = 1.1 M), the largest H2 evolution activity was attained with AscH− (0.30 M) and AscH2 (0.80 M) (Fig. S2 in ESI†). The smaller concentration of AscH− results in less efficient reductive quenching of the [Ru(bpy)3]2+* emission (* denotes the excited state). The quenching efficiency of [Ru(bpy)3]2+* (Ered = 0.77 V vs. SCE in MeCN)20 by AscH− (0.30 M) with AscH2 (0.80 M) was determined to be 95% (Fig. S3 in ESI†). On the other hand, the smaller concentration of AscH2 may retard H2 production due to decreasing the acidity. When H2O was replaced by D2O, D2 and HD were produced without formation of H2. Thus, hydrogen was produced from water and ascorbic acid as electron and proton sources. The observed deuterium kinetic isotope effect (KIE) in Fig. 1 (kH/kD = 1.8 in the initial stage) suggests that the Co–H bond cleavage of a cobalt hydride intermediate ([CoIII(H)(Ch)]) by proton may be the rate-determining step for the photocatalytic H2 evolution (vide infra).
:1 v/v) of [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) containing ascorbic acid (AscH2) and ascorbate (AscH−) (Eox = 0.43 V vs. SCE) as an electron donor and CoII(Ch) (Ered = −0.96 V vs. SCE) (Fig. S1 in the ESI†) as a catalyst resulted in H2 evolution (Fig. 1, black line). When the ratio of AscH− to AscH2 was changed as fixed total concentrations of AscH2 and AscH− ([AscH2] + [AscH−] = 1.1 M), the largest H2 evolution activity was attained with AscH− (0.30 M) and AscH2 (0.80 M) (Fig. S2 in ESI†). The smaller concentration of AscH− results in less efficient reductive quenching of the [Ru(bpy)3]2+* emission (* denotes the excited state). The quenching efficiency of [Ru(bpy)3]2+* (Ered = 0.77 V vs. SCE in MeCN)20 by AscH− (0.30 M) with AscH2 (0.80 M) was determined to be 95% (Fig. S3 in ESI†). On the other hand, the smaller concentration of AscH2 may retard H2 production due to decreasing the acidity. When H2O was replaced by D2O, D2 and HD were produced without formation of H2. Thus, hydrogen was produced from water and ascorbic acid as electron and proton sources. The observed deuterium kinetic isotope effect (KIE) in Fig. 1 (kH/kD = 1.8 in the initial stage) suggests that the Co–H bond cleavage of a cobalt hydride intermediate ([CoIII(H)(Ch)]) by proton may be the rate-determining step for the photocatalytic H2 evolution (vide infra).
 | ||
| Fig. 1 Time courses of H2 evolution in the photocatalytic reduction of proton in an Ar-saturated H2O/MeCN (black) and D2O/MeCN (red) mixed solution (1:1 v/v) containing [RuII(bpy)3]2+ (2.0 mM), AscH2 (0.80 M), AscHNa (0.30 M) and CoII(Ch) (25 μM) under irradiation of visible light (λ > 420 nm) at 298 K. | ||
The concentration of CoII(Ch) was optimised to be 50 μM for the efficient photocatalytic H2 evolution. The absorption of [Ru(bpy)3]2+ is blocked by the larger concentration of CoII(Ch) (Fig. S4 in ESI†).21 The quantum yield of the photocatalytic H2 evolution was determined to be 12% using a ferric oxalate actinometer (see the Experimental section in ESI†). This value is similar to the highest value reported for photocatalytic H2 evolution using a cobalt terpyridine complex (Φ = 0.13).22
Nanosecond transient absorption spectra of an H2O/MeCN solution of [Ru(bpy)3]2+ with AscH2 and AscH− are shown in Fig. 2, where appearance of the absorption band at 500 nm due to [Ru(bpy)3]+ is observed upon the nanosecond laser excitation. Thus, electron transfer from AscH− to [Ru(bpy)3]2+* occurred to produce AscH˙ and [Ru(bpy)3]+. The rate constant of electron transfer from AscH− to [Ru(bpy)3]2+* (ket) was determined to be 8.0 × 108 M−1 s−1 from a slope of Stern–Volmer plot (KSV = 3.5 × 102 M−1) and the lifetime of [Ru(bpy)3]2+* (0.44 μs in water/MeCN 1:1 v/v) (Fig. S5 in ESI†).23 The decay rate of absorbance at 500 nm due to [Ru(bpy)3]+ obeyed the second-order kinetics of bimolecular back electron transfer from [Ru(bpy)3]+ to AscH˙. In the presence of CoII(Ch), the decay of absorbance became much faster because of electron transfer from [Ru(bpy)3]+ to CoII(Ch) as shown in Fig. 2b. The decay rate constant linearly increased with increasing the concentration of [CoII(Ch)] (Fig. S6 in ESI†). The rate constant of electron transfer from [Ru(bpy)3]+ to CoII(Ch) was determined to be 2.5 × 109 M−1 s−1 from the slope of dependence of the first-order decay rate constant on concentration of CoII(Ch) (Fig. S6b in ESI†).
 | ||
| Fig. 2 (a) Transient absorption spectra after laser excitation (λ = 450 nm) of [RuII(bpy)3]2+ (80 μM) in the presence of AscH2 (0.80 M) and AscHNa (0.30 M) in a deaerated H2O/MeCN mixed solution (1:1 v/v) at 298 K. (b) Time profiles of absorbance at 500 nm due to decay of [Ru(bpy)3]+ in the presence of various concentrations of CoII(Ch) (0–20 μM) in deaerated H2O/MeCN mixed solutions (1:1 v/v) containing [RuII(bpy)3]2+ (80 μM), AscH2 (0.80 M), AscHNa (0.30 M). | ||
To examine the reaction of [CoI(Ch)]− that is produced by electron transfer from [Ru(bpy)3]+ to CoII(Ch), [CoI(Ch)]− was prepared by the one-electron reduction of CoII(Ch) by decamethylcobaltocene [Co(Cp*)2] in MeCN (Fig. S7 in ESI†). The UV-vis absorption band of [CoI(Ch)]− (red line in Fig. 3a; λmax = 510 nm) decreased with increasing absorption band at 660 nm (black line) at 66 ms after addition of acetic acid (CH3COOH) (0.30 M). Then, this absorption band was finally blue shifted to λmax = 652 nm, which is due to [CoIII(Ch)]+.24,25 Thus, [CoI(Ch)]− may react with CH3COOH to form the hydride complex ([CoIII(H)(Ch)(CH3COO)]−: λmax = 660 nm), from which H2 was evolved by the reaction with CH3COOH to produce [CoIII(Ch)]+. The reaction of [CoI(Ch)]− with CH3COOH was monitored by the absorption change at 652 nm due to [CoIII(Ch)]+ as shown in Fig. 3, where the rate of the formation of [CoIII(Ch)]+ obeyed first-order kinetics (Fig. S8 in ESI†). The first-order rate constant increased with increasing concentration of CH3COOH to approach a constant value (Fig. 3b). Such a saturation behaviour indicates that CH3COOH is not involved in the rate-determining step and that the reaction of [CoI(Ch)]− with CH3COOH proceeds via formation of the hydride complex ([CoIII(H)(Ch)(CH3COO)]−), followed by the rate-determining heterolytic cleavage of the CoIII–H bond. The subsequent reaction of the released hydride ion with CH3COOH to produce H2 and [CoIII(Ch)]+ may be fast as compared with the back reaction of the CoIII–H bond cleavage (Scheme 1). The kinetic equation for the formation of [CoIII(Ch)]+ is given by eqn (1),
| d[[CoIII(Ch)]+]/dt = k[[CoIII(H)(Ch)(CH3COO)]−] | (1) |
| [[CoIII(H)(Ch)(CH3COO)]−] = K[CH3COOH]([[CoI(Ch)]−]0 − [[CoIII(Ch)]+])/(1 + K[CH3COOH]) | (2) |
| d[[CoIII(Ch)]+]/dt = kK[CH3COOH]([[CoI(Ch)]−]0 − [[CoIII(Ch)]+])/(1 + K[CH3COOH]) | (3) |
| kobs = kK[CH3COOH]/(1 + K[CH3COOH]) | (4) |
| kobs−1 = 1/kK·[CH3COOH]−1 + 1/k | (5) |
 | ||
| Fig. 3 (a) UV-vis absorption spectral changes of [CoI(Ch)]− (30 μM) upon addition of CH3COOH (0.30 M) in dearated MeCN at 298 K. The black and blue lines show the spectra taken at 66 ms and 1 s after mixing, respectively. The red line shows UV-vis absorption spectrum of [CoI(Ch)]− (15 μM) formed by the electron-transfer reduction of CoII(Ch) (15 μM) with CoCp2* (300 μM) in dearated MeCN at 298 K. (b) Plot of kobs for the rate of formation of [CoIII(Ch)]+vs. [CH3COOH]. | ||
 | ||
| Scheme 1 Mechanism of hydrogen formation by the reaction of [CoI(Ch)]− with CH3COOH. | ||
When CH3COOH was replaced by CH3COOD, the deuterium kinetic isotope effect (KIE) was observed (Fig. S10 in ESI†),26 indicating that the cleavage of the Co–H bond of [CoIII(H)(Ch)(CH3COO)]− or O–H bond of CH3COOH is involved in the rate-determining step of the reaction of [CoI(Ch)]− with CH3COOH. Because CH3COOH is not involved in the rate-determining step (vide infra), the cleavage of the Co–H bond of [CoIII(H)(Ch)(CH3COO)]− is the rate-determining step of the reaction of [CoI(Ch)]− with CH3COOH. The KIE value was 1.7 which is virtually the same as observed for the photocatalytic H2 evolution (KIE = 1.8, Fig. 1), indicating that the heterolytic Co–H bond cleavage of [CoIII(H)(Ch)(CH3COO)]− is also the rate-determining step in the photocatalytic H2 evolution.
[CoIII(Ch)]+ produced by the reaction of [CoIII(H)(Ch)–(CH3COO)]− with CH3COOH is reduced by AscH− to form CoII(Ch) as shown by stopped-flow measurements in Fig. 4.27 The rate constant of electron transfer from AscH− to [CoIII(Ch)]+ that was prepared by the one-electron oxidation of CoII(Ch) with (p-BrC6H4)3N˙+SbCl6− in H2O/MeCN was determined to be 1.5 × 103 M−1 s−1 from the linear dependence of the first-order rate constant on concentration of AscH− (Fig. S11 in ESI†).
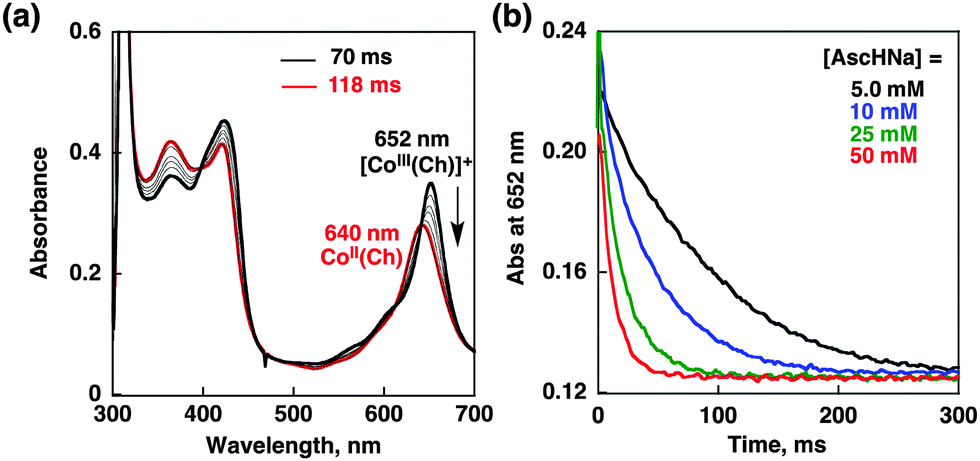 | ||
| Fig. 4 (a) UV-vis absorption spectral changes in the electron-transfer reduction of [CoIII(Ch)]+ (15 μM) with AscHNa (50 mM) in air-saturated H2O/MeCN mixed solutions (1:1 v/v) at 298 K taken at 70 ms and 118 ms after mixing. (b) Decay time profiles of absorbance at 652 nm due to [CoIII(Ch)]+ in the presence of various concentrations of AscHNa in air-saturated H2O/MeCN mixed solutions (1:1 v/v) at 298 K. | ||
The photocatalytic cycle is summarized in Scheme 2. Photoexcitation of [Ru(bpy)3]2+ resulted in electron transfer from AscH− to [Ru(bpy)3]2+* to produce [Ru(bpy)3]+, followed by electron transfer from [Ru(bpy)3]+ to CoII(Ch) to produce [CoI(Ch)]−, which reacts with AscH2 to produce [CoIII(H)(Ch)(AscH)]−. Hydrogen is generated by the reaction of [CoIII(H)(Ch)(AscH)]− with AscH2via the Co–H bond heterolysis to produce [CoIII(Ch)]+,28,29 which is reduced by AscH− to regenerate CoII(Ch). In such a case, a one-photon two-electron process is made possible, because one photon is required to produce [CoI(Ch)]− for H2 evolution and another electron is provided thermally by AscH−.
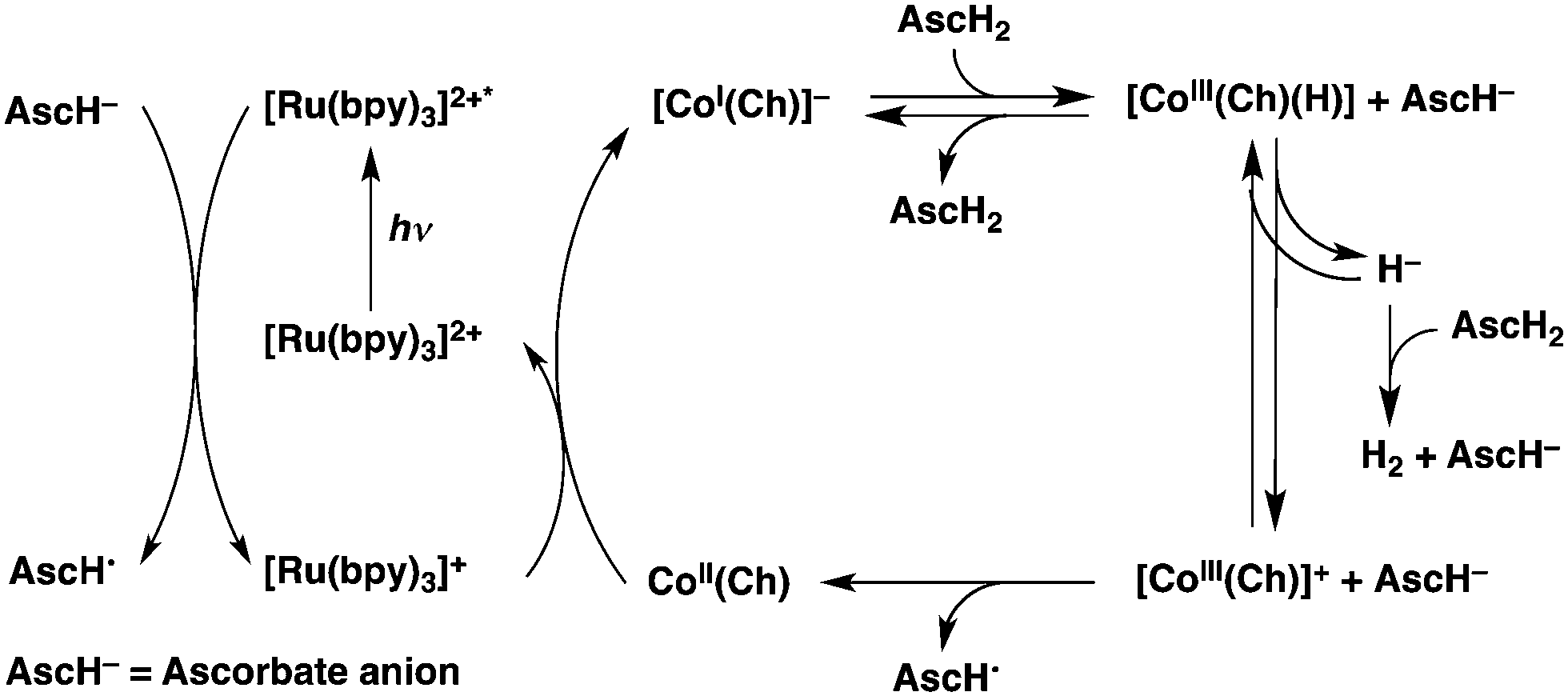 | ||
| Scheme 2 Mechanism of photocatalytic hydrogen evolution from AscH− and AscH2 with [Ru(bpy)3]2+ and CoII(Ch). | ||
In conclusion, CoII(Ch) acts as an efficient catalyst for photocatalytic H2 evolution from ascorbic acid with [Ru(bpy)3]2+ as a photocatalyst to attain the high quantum yield via a one-photon two-electron process in which the second electron is provided thermally from ascorbic acid.
This work was supported by Grants-in-Aid (no. 26620154 and 26288037 to K.O.) and JSPS fellowship (No. 25-727 to K.M.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); ALCA and SENTAN projects from JST, Japan (to S.F.).
Notes and references
- R. A. Kerr and R. F. Service, Science, 2005, 309, 101 CrossRef CAS PubMed.
- X. Song, H. Wen, C. Ma, H. Chen and C. Chen, New J. Chem., 2015, 39, 1734 RSC.
- K. Kawano, K. Yamauchi and K. Sakai, Chem. Commun., 2014, 50, 9872 RSC.
- X. Wang, S. Goeb, Z. Ji, N. A. Pogulaichenko and F. N. Castellano, Inorg. Chem., 2011, 50, 705 CrossRef CAS PubMed.
- L.-Z. Fu, L.-L. Zhou, L.-Z. Tang, Y.-X. Xhang and S.-Z. Zhan, J. Power Sources, 2015, 280, 453 CrossRef CAS.
- S. Fukuzumi, Curr. Opin. Chem. Biol., 2015, 25, 18 CrossRef CAS PubMed.
- D. Basu, S. Mazumder, X. Shi, H. Baydoun, J. Niklas, O. Poluektov, H. B. Schlegel and C. N. Verani, Angew. Chem., Int. Ed., 2015, 54, 2105 CrossRef CAS PubMed.
- A. Call, Z. Codola, F. Acuna-Pares and J. Lloret-Fillol, Chem. – Eur. J., 2014, 20, 6171 CrossRef CAS PubMed.
- H. Lv, W. Guo, K. Wu, Z. Chen, J. Bacsa, D. G. Musaev, Y. V. Geletii, S. M. Lauinger, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 14015 CrossRef CAS PubMed.
- L. Chen, G. Chen, C.-F. Leung, S.-M. Yiu, C.-C. Ko, E. Anxolabéhère-Mallart, M. Robert and T.-C. Lau, ACS Catal., 2015, 5, 356 CrossRef CAS.
- A. Zarkadoulas, E. Koutsouri, C. Kefalidi and C. A. Mitsopoulou, Coord. Chem. Rev., 2014, 11, 6 Search PubMed.
- K. Maeda, M. Eguchi and T. Oshima, Angew. Chem., Int. Ed., 2014, 53, 13164 CrossRef CAS PubMed.
- J. Zhao, Y. Ding, J. Wei, X. Du, Y. Yu and R. Han, Int. J. Hydrogen Energy, 2014, 39, 18908 CrossRef CAS.
- E. Deponti, A. Luisa, M. Natali, E. lengo and F. Scandola, Dalton Trans., 2014, 43, 16345 RSC.
- H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227 RSC.
- A. Rodenberg, M. Orazietti, B. Probst, C. Bachmann, R. Alberto, K. K. Baldridge and P. Hamm, Inorg. Chem., 2015, 54, 646 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, Angew. Chem., Int. Ed., 2011, 50, 728 CrossRef CAS PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995 CrossRef CAS PubMed.
- K. Mase, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 2800 CrossRef CAS PubMed.
- J. Yuasa and S. Fukuzumi, J. Am. Chem. Soc., 2006, 128, 14281 CrossRef CAS PubMed.
- The photocatalytic H2 evolution in our optimized conditions, the absorption at λ = 450 nm of [Ru(bpy)3]2+ (2.0 mM, ε450nm = 1.4 × 104 M−1 cm−1) is not significantly blocked by that of CoII(Ch) (Abs450nm = 0.38; ε450nm = 1.8 × 104 M−1 cm−1) under this experimental conditions.
- C. V. Krishnan and N. Sutin, J. Am. Chem. Soc., 1981, 103, 2141 CrossRef CAS.
- The emission lifetime of [Ru(bpy)3]2+* in water at 298 K was reported to be 0.58 μs; see: J. V. Houten and R. J. Watts, J. Am. Chem. Soc., 1976, 98, 4853 CrossRef.
- The spectrum of [CoIII(Ch)]+ obtained by the reaction [CoI(Ch)]− with CH3COOH was identical to that of [CoIII(Ch)]+ prepared by the electron-transfer oxidation of CoII(Ch) by a one-electron oxidizing reagent of (p-BrC6H4)3N˙+SbCl6− (Ered = 1.05 V vs. SCE).19.
- [CoIII(Ch)]+ or CoIII(H)(Ch) species is not re-reduced by large excess of Co(Cp*)2, under the present reaction conditions because CoII(Ch) with 20 molar equiv. of Co(Cp*)2 is necessary to quantitatively produce [CoI(Ch)]− as shown in ESI,† Fig. S7. Co(Cp*)2 (E1/2+/0 = −1.47 V vs. SCE) is unstable even in carefully degassed and dehydrated MeCN.
- The KIE value was determined from the kobs values at [CH3COOH] = [CH3COOD] = 1.0 M.
- Neither oxidation of [CoIII(Ch)]+ nor O2 reduction was observed under the basic reaction conditions.
- S. Mandal, S. Shikano, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 15294 CrossRef CAS PubMed.
- No Co(II)–H complex is involved in the heterolysis of the Co–H bond as reported in ref. 28.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and cyclic voltammograms (Fig. S1), time courses of H2 evolution (Fig. S2), emission spectra (Fig. S3 and S5), UV-vis absorption spectra (Fig. S4 and S7) and kinetic data (Fig. S6–S11). See DOI: 10.1039/c5cc05064b |
| This journal is © The Royal Society of Chemistry 2015 |