
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomochiral metal phosphonate nanotubes†
Xun-Gao
Liu‡
a,
Song-Song
Bao‡
a,
Jian
Huang
a,
Kazuya
Otsubo
b,
Jian-Shen
Feng
a,
Min
Ren
a,
Feng-Chun
Hu
c,
Zhihu
Sun
c,
Li-Min
Zheng
*a,
Shiqiang
Wei
*c and
Hiroshi
Kitagawa
*b
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, China. E-mail: lmzheng@nju.edu.cn
bDivision of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-Oiwakecho, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: kitagawa@kuchem.kyoto-u.ac.jp
cNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029, P. R. China. E-mail: sqwei@ustc.edu.cn
First published on 20th August 2015
Abstract
A new type of homochiral metal–organic nanotubular structures based on metal phosphonates are reported, namely, (R)- or (S)-[M(pemp)(H2O)2][M = CoII (1), NiII (2)] [pemp2− = (R)- or (S)-(1-phenylethylamino)methylphosphonate]. In these compounds, the tube-walls are purely inorganic, composed of metal ions and O–P–O bridges. The cavity of the nanotube is hydrophilic with one coordination water pointing towards the center, while the outer periphery of the nanotube is hydrophobic, decorated by the phenylethyl groups of pemp2−. The thermal stabilities, adsorption and proton conductivity properties are investigated.
Since the discovery of carbon nanotubes by Iijima in 1991,1 nanotubular materials have received considerable attention due to their unique physical and chemical properties and potential applications including catalysis, drug delivery, energy storage and sensors.2 The fabrication of nanotubular materials is also very important for understanding ion or water transport within complex biological channels.3 So far a number of pure inorganic or organic nanotubular materials have been constructed successfully through rolling-up or supramolecular approaches.4,5 In contrast, there are much fewer metal–organic hybrid systems showing discrete or one-dimensional (1D) tubular structures.6–10 Most of the work focused on the structures, while their functions remain largely unexplored.9
On the other hand, homochiral structures are ubiquitous in nature. The combination of chirality with porosity can result in materials that have potential applications in enantioselective separation and catalysis, nonlinear optics and multifunctional materials.11 As far as we are aware, homochiral zeolites still remain a synthetic challenge with only one exception of pure chiral silica.12 Homochiral metal–organic frameworks (MOFs) are more common than chiral zeolites,11,13 but those with tubular architectures are extremely rare.10 Metal phosphonates as an important class of metal–organic hybrid materials can be good candidates for the construction of porous materials with high thermal and water stabilities.14,15 Despite the increasing number of metal phosphonates reported in the past two decades, however, only two uranyl compounds exhibit nanotubular structures.16 Homochiral metal phosphonates are rare,17,18 and those with tubular structures have never been documented.
In this paper, we report the first examples of homochiral metal phosphonate nanotubes, namely, (R)-[Co(pemp)(H2O)2] (R-1), (S)-[Co(pemp)(H2O)2] (S-1), (R)-[Ni(pemp)(H2O)2] (R-2) and (S)-[Ni(pemp)(H2O)2] (S-2) [pemp2− = (R)- or (S)-(1-phenylethylamino)methylphosphonate]. They provide a new type of chiral nanotubes among the metal–organic hybrid materials. The compounds can be dehydrated and rehydrated while maintaining their chiral tubular nature, as confirmed by the powder XRD, CD and XAFS measurements. The adsorption and proton conductivity properties of these compounds are also investigated.
Compounds R-1, S-1, R-2 and S-2 were synthesized through hydrothermal reactions of cobalt or nickel salt and pempH2 at 140 °C for 4 days. These compounds are isomorphous according to their powder XRD patterns (Fig. S3–S6, ESI†). The R- and S-isomers are a pair of enantiomers. Single-crystal structural determination reveals that all crystallize in the hexagonal chiral space group P63 (Table S1, ESI†).§ Take R-1 as an example. The asymmetric unit contains one independent CoII ion, one R-pemp2− ligand and two coordinated water molecules (Fig. 1a, left). Each CoII ion adopts a slightly distorted octahedral coordination geometry, surrounded by three phosphonate oxygen (O1, O2A, O3B) atoms and one nitrogen (N1) atom from three equivalent R-pemp2− ligands and two water molecules [Co1–O: 2.070(4)–2.180(3) Å, Co1–N: 2.235(5) Å]. The enantiopure R-pemp2− chelates the Co atom through N1 and O1 atoms, and bridges the equivalent Co atoms via the other two phosphonate oxygens (O2, O3), hence inducing the chirality on the metal centers with Λ-configuration in R-1. Each {PO3C} tetrahedron is corner-sharing with three {CoO5N} octahedra through three phosphonate oxygen atoms and vice versa, forming an infinite chiral nanotube running along the 63 screw axis in the c-direction (Fig. 1b, left). The Co⋯Co distances over the O–P–O bridges are 4.998(1) Å and 6.169(1) Å. One of the two coordinated water molecules (O2W) point to the center of the window, and form hydrogen bonds with the phosphonate oxygen atoms [O2W⋯O1: 2.928(5) Å]. Thus the cavity of the nanotube is hydrophilic, while the outer periphery of the nanotube is hydrophobic decorated by the phenylethyl groups of R-pemp2−. The diameter of the nanotube, defined by the distance between the opposite coordinated water oxygen chains, is ca. 3.76 Å (3.01 Å between opposite water hydrogen chains) (van der Waals radii not taken into account), similar to those in a 3D compound Zn(O3PCH2OH).19 The distance of opposite cobalt chains is ca. 7.86 Å (van der Waals radii not taken into account), which defines approximately the inner diameter after removal of water. The nanotubes are packed in a hexagonal arrangement, where van der Waals interactions are dominant between the phenyl rings of the adjacent tubes (Fig. 1c, left). The building unit, inorganic tube and packing diagram of compound S-1 are also shown in Fig. 1a–c (right) for comparison.
 | ||
| Fig. 1 (a) Building units of structures R-1 (left) and S-1 (right); (b) the inorganic tubes in structures R-1 (left) and S-1 (right) viewed along the c-axis. The coordination water are highlighted in yellow; (c) structures R-1 (left) and S-1 (right) packed along the c-axis. (d) Hydrogen bond interactions inside the nanotube of R-1. | ||
Thermal analyses show that compounds 1 and 2 experience the first-step weight losses in the range 100–170 °C for 1 (10.9% for R-1, 11.6% for S-1) and 120–200 °C for 2 (11.4% for R-2 and S-2), respectively, corresponding to the removal of two coordinated water molecules (calcd 11.7% for 1 and 2) (Fig. S7, ESI†). This is followed by a distinct plateau before a second weight loss step occurs above ca. 280 °C, attributed to the combustion of the organic constituents and the collapse of the structure. The dehydration process is accompanied by a color change from purple red to blue for 1, and green to yellow for 2. The XRD measurements on R-1-de and R-2-de, obtained by thermal treatment at 180 °C (for R-1) or 200 °C (for R-2) for two hours, reveal that the nanotubular structures are maintained after removing the coordination water. For R-1, the immersion of the dehydrated samples in water leads to the recovery of the original phases (Fig. S10, ESI†). While for R-2, the dehydration/rehydration process is not reversible at room temperature (Fig. S11, ESI†).
Solid-state circular dichroism (CD) spectra were obtained for enantio-isomers of 1 and 2. As shown in Fig. 2a, negative Cotton effects of R-1 are evident at 258 nm and positive dichroic signals are centered at 471, 489 and 556 nm, while S-1 exhibits the opposite sign of Cotton effects at the same wavelength. Compounds R-2 and S-2 exhibit mirror dichroic signals at ca. 261, 418 and 670 nm (Fig. 2b). These results further prove the enantiomeric nature of these compounds, with the optical activities originating from both the chiral phosphonate ligands and the metal centers. The chiral nature remains after dehydration, as confirmed by the presence of CD signals at ca. 262 nm. Interestingly, the peaks in the visible region disappear in all four cases, indicating that the chirality in the metal centers is not retained after dehydration. The rehydration of R-1-de and S-1-de leads to the recovery of the CD signals in the UV-vis region. In contrast, the immersion of samples R-2-de and S-2-de in water does not change their CD spectra. The results demonstrate again that the dehydration/rehydration process is reversible for compounds R-1 and S-1, while not for compounds R-2 and S-2. Furthermore, the second harmonic generation (SHG) measurements were carried out for the four compounds before and after dehydration. All exhibit SHG responses about 0.4 or 0.3 times that of urea.
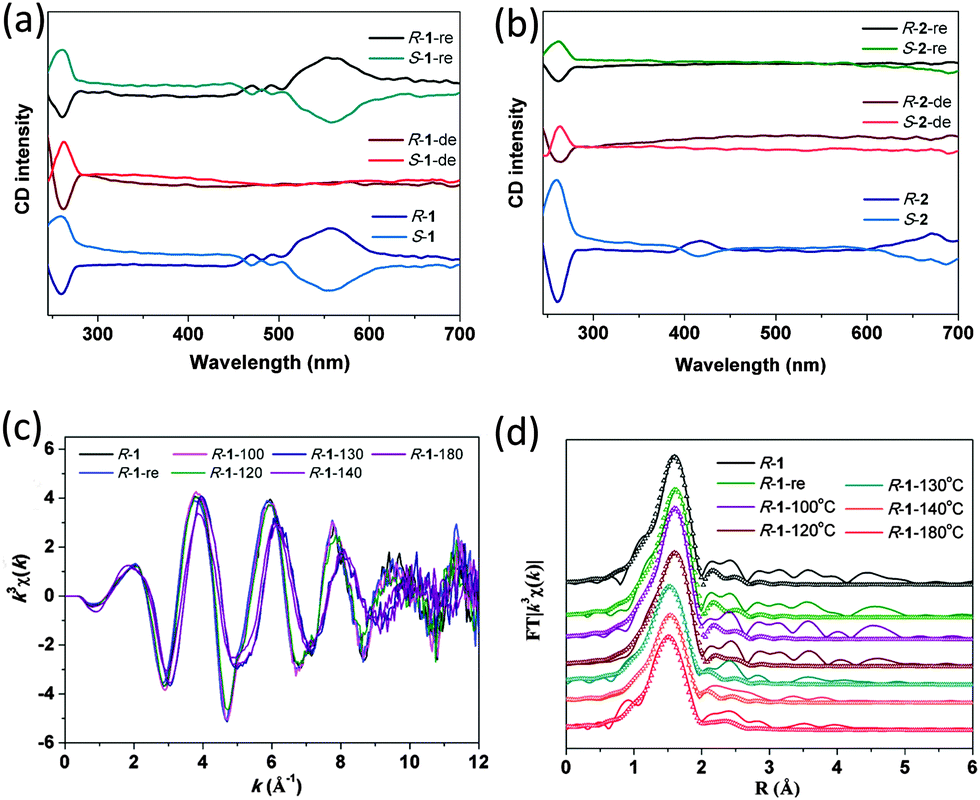 | ||
| Fig. 2 Solid-state circular dichroism spectra for (a) compounds R-1 and S-1, and (b) R-2 and S-2 before and after dehydration and rehydration. (c) Co-K edge k3-weight EXAFS signals taken at RT for compound R-1 at different dehydration temperatures. (d) Fourier transformed space (R space) and their fitting curves (dotted lines) for R-1 at Co K-edge. | ||
To monitor the change in the local coordination sphere of the metal ions after dehydration, Co(Ni) K-edge EXAFS spectroscopic measurements20 were conducted for R-1 and R-2 as well as their dehydrated samples pretreated at different temperatures for 1 h (Fig. S13 and S14, ESI†). Curve-fitting was performed for the first FT peak in the R-space using Hanning windows (for R-1) or Kaiser–Bessel windows (for R-2) by taking into account the Co–O(N) or Ni–O(N) coordination pairs (Fig. 2c and d and Fig. S15, S16, ESI†). The extracted structural parameters are summarized in Tables S5 and S6 (ESI†). For R-1, the data fit leads to five oxygen atoms at 2.07 Å and one nitrogen atom at 2.25 Å, which are close to those obtained by the single crystal structural determination (av. 2.117 and 2.236 Å, respectively). Upon heating, both the Co–O(N) bond lengths and the coordination number of cobalt remain almost the same up to 120 °C. Significant structural change is observed when the sample is thermally treated at 130 °C, where the CoII becomes four-coordinated and the Co–O(N) distances are shortened (Co–O: 1.98, Co–N: 2.14 Å). The coordination environment of cobalt in the fully dehydrated sample R-1-180 °C is similar to that in R-1-130 °C. Rehydration of the sample R-1-180 °C by immersing in water leads to the recovery of R-1, evidenced by the Co–O(N) distances and coordination number of six (Table S5, ESI†).
For R-2, the data fit leads to five oxygen atoms at 2.05 Å and one nitrogen atom at 2.15 Å, which are close to those obtained by the single crystal structure determination (av. 2.066 and 2.166 Å). In the fully dehydrated sample R-2-200 °C, the coordination number of nickel is still six, and the Ni–O(N) distances remain almost the same (Table S6, ESI†). The result suggests that the vacant sites of the nickel ion after dehydration could be refilled with the neighboring phosphonate oxygen atoms, which may explain the irreversible dehydration/rehydration process for R-2 at room temperature. The EXAFS results are in agreement with the TG and XRD data.
Water adsorption and desorption isotherms were collected at 25 °C on the dehydrated samples of R-1 and R-2, pre-treated at 100 °C under vacuum for 12 hours. As shown in Fig. 3 and Fig. S17 (ESI†), both experience a one-step adsorption process. The amount of water adsorbed at P/P0 = 0.98 is 2.94 (for R-1-de) and 2.79 (for R-2-de) molecules of water per formula unit. Interestingly, the desorption isotherms display significant hysteresis in both cases and show a gradual release of water molecules upon decreasing the relative pressure. The number of water molecules per formula unit at P/P0 = 0.02 is 1.98 for the cobalt sample and 1.31 for the nickel sample. The former agrees well with the two coordinated water molecules in compound R-1, indicating that each cobalt ion re-binds two water molecules which cannot be removed at 25 °C. The latter is much less than two coordination water molecules expected for R-2, again suggesting that the water molecules do not re-coordinate to the nickel ions after dehydration. We have repeated the water adsorption and desorption isotherms of both R-1 and R-2 three times without significant change in the curves.
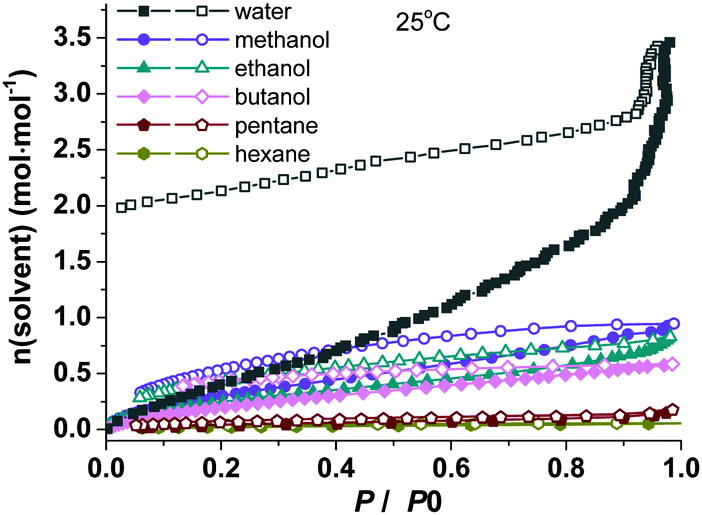 | ||
| Fig. 3 The solvent adsorption (filled points) and desorption (open points) isotherms for compound R-1 at 25 °C. | ||
The adsorption behavior of organic molecules such as alcohols and alkanes was also examined for the dehydrated samples of R-1 and R-2. Fig. 3 shows the adsorption and desorption isotherms of R-1-de at 25 °C. The amounts adsorbed at P/P0 = 0.98 are 1.15 molecules of methanol, 0.96 molecules of ethanol and 0.84 molecules of butanol per formula unit, which are significantly less compared with that of water (2.94 mol mol−1). In addition, the adsorption towards pentane and hexane is nearly zero. Similar behavior is observed for sample R-2-de (Fig. S17, ESI†). Noting that the kinetic diameters of water (2.7 Å) is smaller than those of methanol (3.6 Å), ethanol (4.5 Å), butanol (5.0 Å) and the alkane molecules (4.3–4.5 Å),21 the highly selective water adsorption capabilities of R-1-de and R-2-de over alcohols and alkanes are attributed to the small diameter (7.86 Å) and also to the hydrophilic nature inside the nanotubes.
The N2 and H2 gas adsorption and desorption isotherms were also measured at 77 K. The BET surface areas obtained from the N2 sorption isotherms are 10.2 and 12.0 m2 g−1 for R-1-de and R-2-de, respectively. As shown in Fig. S18 (ESI†), compound R-2-de can adsorb more H2 than R-1-de at 77 K (10.4 vs. 3.56 cm3 g−1 at 100 kPa), the origin of which is not clear to us. Distinct hysteresis is observed in the H2 desorption isotherms in both cases. Apparently, the release of H2 molecules is very slow upon decreasing the pressure. The results are in agreement with the presence of nanotubes with small inner diameters.
In order to study the proton transportation capabilities of these nanotubular materials, we initially measured the proton conductivities of powder pellets of R-1 and R-2 by a quasi-four probe method using a Solartron SI 1260 impedance/gain-phase analyzer with a 1296 dielectric interface. The conductivities were determined from the semicircular Nyquist plots at different temperatures and relative humidities (RHs). It is found that the proton conductivities are 2.16 × 10−6 S cm−1 for R-1 and 1.61 × 10−6 S cm−1 for R-2 at 25 °C and 95% RH. The values are highly humidity dependent, and drop to 1.21 × 10−11 and 6.50 × 10−12 S cm−1 for the two samples at 25 °C and 50% RH (Fig. S21 and S22, ESI†). The temperature dependence of the proton conductivities was measured in the range of 15–45 °C at 95% RH for the two samples (Fig. S23–S26, ESI†). The activation energies (Ea) are estimated to be 0.30 and 0.24 eV (2nd cycle) for R-1 and R-2, respectively, suggesting a Grotthuss-type conduction mechanism.22
Noting that compounds R-1 and R-2 are neutral, both the proton source and carrier are likely coordinated water, where the acidity is enhanced by coordination to the metal ions.23 However, the conductivities of R-1 and R-2 are much lower compared with the other phosphonate-based MOFs.24 To check whether the conductivity originates from the proton transportation inside the nanotube, we performed the measurements using a single crystal of R-1 with dimensions 1.3 × 0.1 × 0.07 mm3. It is found that the conductivity along the tube direction is ca. 6.7 × 10−11 S cm−1 at 25 °C and 95% RH (Fig. S29, ESI†). Obviously, the overall proton conductivity of the powder samples mainly arises from the boundary effects. The result is in agreement with the structures of R-1 and R-2, where the hydrogen-bonded six-member rings constructed by three O2W and three O1 atoms are not connected to neighboring rings through hydrogen bonds (Fig. 1d), thus blocking an efficient proton conducting pathway within the hydrophilic channels.
In summary, we have succeeded in obtaining the first examples of homochiral nanotubular metal phosphonates, namely, (R)- or (S)-[M(pemp)(H2O)2] (M = CoII, NiII). The fully dehydrated samples exhibit highly selective water adsorption capabilities over the alcohol and alkane molecules. Further work is in progress to understand the formation mechanism of these nanotubes and to explore new tubular metal phosphonates with expanded inner sizes.
This work is supported by the National Basic Research Program of China (2013CB922102) and the NSF of China (No. 21371094, 11135008, 11305172). We thank the State Key Laboratory of Structure Chemistry, Fujian Institute of Research on the Structure of Matter (China), for the single crystal structural determination. We also thank Prof. Hui Zhang in Xiamen University for valuable suggestions in the CD measurements.
Notes and references
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS PubMed.
- (a) W. Xiong, F. Du, Y. Liu, A. Perez Jr., M. Supp, T. S. Ramakrishnan, L. Dai and L. Jiang, J. Am. Chem. Soc., 2010, 132, 15839–15841 CrossRef CAS PubMed; (b) V. Biju, Chem. Soc. Rev., 2014, 43, 744–764 RSC; (c) H. G. Park and Y. Jung, Chem. Soc. Rev., 2014, 43, 565–576 RSC.
- (a) H. X. Sui, B. G. Han, J. K. Lee, P. Walian and B. K. Jap, Nature, 2001, 414, 872–878 CrossRef CAS PubMed; (b) D. Fu, A. Libson, L. J. Miercke, C. Weitzman, P. Nollert, J. Krucinski and R. M. Stroud, Science, 2000, 290, 481–486 CrossRef CAS.
- C. N. R. Rao, S. R. C. Vivekchand, K. Biswas and A. Govindaraj, Dalton Trans., 2007, 3728–3749 RSC.
- (a) D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS; (b) V. G. Organo and D. M. Rudkevich, Chem. Commun., 2007, 3891–3899 RSC.
- P. Thanasekaran, T.-T. Luo, C.-H. Lee and K.-L. Lu, J. Mater. Chem., 2011, 21, 13140–13149 RSC and references therein.
- G. Wu, J. Bai, Y. Jiang, G. Li, J. Huang, Y. Li, C. E. Anson, A. K. Powell and S. Qiu, J. Am. Chem. Soc., 2013, 135, 18276–18279 CrossRef CAS.
- K. Otsubo, Y. Wakabayashi, J. Ohara, S. Yamamoto, H. Matsuzaki, H. Okamoto, K. Nitta, T. Uruga and H. Kitagawa, Nat. Mater., 2011, 10, 291–295 CrossRef CAS PubMed.
- (a) T. Panda, T. Kunduz and R. Banerjee, Chem. Commun., 2012, 48, 5464–5466 RSC; (b) D. K. Unruh, K. Gojdas, A. Libo and T. Z. Forbes, J. Am. Chem. Soc., 2013, 135, 7398–7401 CrossRef CAS PubMed; (c) P. Neuhaus, A. Cnossen, J. Q. Gong, L. M. Herz and H. L. Anderson, Angew. Chem., Int. Ed., 2015, 54, 7344–7348 CrossRef CAS PubMed.
- (a) G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049–1052 CrossRef CAS; (b) H.-T. Zhang, Y.-Z. Li, T.-W. Wang, E. N. Nfor, H.-Q. Wang and X.-Z. You, Eur. J. Inorg. Chem., 2006, 3532–3536 CrossRef CAS PubMed.
- (a) H. K. Ngo and W. B. Lin, Top. Catal., 2005, 34, 85–92 CrossRef CAS; (b) Y. Liu, W. M. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112–4135 CrossRef CAS PubMed; (c) P. Peluso, V. Mamane and S. Cossu, J. Chromatogr. A, 2014, 1363, 11–26 CrossRef CAS PubMed; (d) J. Crassous, Chem. Soc. Rev., 2009, 38, 830 RSC; (e) C. Train, M. Gruselle and M. Verdaguer, Chem. Soc. Rev., 2011, 40, 3297 RSC.
- (a) M. E. Davis, Chem. Mater., 2014, 26, 239–245 CrossRef CAS; (b) A. Rojas and M. A. Camblor, Angew. Chem., Int. Ed., 2012, 51, 3854–3856 CrossRef CAS PubMed.
- (a) S.-T. Wu, Z.-W. Cai, Q.-Y. Ye, C.-H. Weng, X.-H. Huang, X.-L. Hu, C.-C. Huang and N.-F. Zhuang, Angew. Chem., Int. Ed., 2014, 53, 12860–12864 CrossRef CAS; (b) X. Zhao, M. Wong, C. Mao, T. X. Trieu, J. Zhang, P. Feng and X. Bu, J. Am. Chem. Soc., 2014, 136, 12572–12575 CrossRef CAS PubMed.
- (a) A. Clearfield, Prog. Inorg. Chem., 1998, 47, 371–510 CrossRef CAS PubMed; (b) A. Clearfield and K. D. Demadis, Metal Phosphonate Chemistry: From Synthesis to Applications, The Royal Society of Chemistry, 2012 Search PubMed.
- (a) G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430–1449 RSC; (b) K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS PubMed; (c) M. T. Wharmby, J. P. S. Mowat, S. P. Thompson and P. A. Wright, J. Am. Chem. Soc., 2011, 133, 1266–1269 CrossRef CAS PubMed; (d) T. Zheng, J. M. Clemente-Juan, J. Ma, L. Dong, S.-S. Bao, J. Huang, E. Coronado and L.-M. Zheng, Chem. – Eur. J., 2013, 19, 16394–16402 CrossRef CAS PubMed.
- (a) D. M. Poojary, D. Grohol and A. Clearfield, Angew. Chem., Int. Ed. Engl., 1995, 34, 1508–1510 CrossRef CAS PubMed; (b) D. M. Poojary, A. Cabeza, M. A. G. Aranda, S. Bruque and A. Clearfield, Inorg. Chem., 1996, 35, 1468–1473 CrossRef CAS.
- (a) H. L. Ngo and W. B. Lin, J. Am. Chem. Soc., 2002, 124, 14298–14299 CrossRef CAS PubMed; (b) Q. Yue, J. Yang, G. H. Li, G. D. Li and J. S. Chen, Inorg. Chem., 2006, 45, 4431–4439 CrossRef CAS PubMed; (c) X. Shi, G. Zhu, S. Qiu, K. Huang, J. Yu and R. Xu, Angew. Chem., Int. Ed., 2004, 43, 6482–6485 CrossRef CAS PubMed.
- (a) X.-G. Liu, S.-S. Bao, Y.-Z. Li and L.-M. Zheng, Inorg. Chem., 2008, 47, 5525–5527 CrossRef CAS PubMed; (b) X.-G. Liu, K. Zhou, J. Dong, C.-J. Zhu, S.-S. Bao and L.-M. Zheng, Inorg. Chem., 2009, 48, 1901–1905 CrossRef CAS PubMed; (c) X.-G. Liu, J. Huang, S.-S. Bao, Y.-Z. Li and L.-M. Zheng, Dalton Trans., 2009, 9837–9842 RSC; (d) T.-T. Wang, S.-S. Bao, J. Huang, Y.-Z. Li and L.-M. Zheng, Dalton Trans., 2013, 42, 1842–1847 RSC; (e) Z.-S. Cai, S.-S. Bao, M. Ren and L.-M. Zheng, Chem. – Eur. J., 2014, 20, 17137–17142 CrossRef CAS PubMed.
- G. B. Hix, B. M. Kariuki, S. Kitchin and M. Tremayne, Inorg. Chem., 2001, 40, 1477–1481 CrossRef CAS PubMed.
- D. C. Koningsberger and R. Prins, X-ray Absorption: Principle, Applications, Techniques of EXAFS, SEXAFS and XANES, Wiley, New York, 1988 Search PubMed.
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836–868 CrossRef CAS.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- N. C. Jeong, B. Samanta, C. Y. Lee, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 51–54 CrossRef CAS PubMed.
- (a) P. Ramaswamy, N. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC and references therein; (b) M. Bazaga-García, R. M. P. Colodrero, M. Papadaki, P. Garczarek, J. Zoń, P. Olivera-Pastor, E. R. Losilla, L. León-Reina, M. A. G. Aranda, D. Choquesillo-Lazarte, K. D. Demadis and A. Cabeza, J. Am. Chem. Soc., 2014, 136, 5731–5739 CrossRef PubMed; (c) S.-S. Bao, K. Otsubo, J. M. Taylor, Z. Jiang, L.-M. Zheng and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 9292–9295 CrossRef CAS PubMed; (d) P. Ramaswamy, N. E. Wong, B. S. Gelfand and G. K. H. Shimizu, J. Am. Chem. Soc., 2015, 137, 7640–7643 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, structural data, SEM, IR, PXRD, TG, additional EXAFS, adsorption/desorption isotherms and proton conductivity figures. CCDC 696828–696830 and 1060995. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc05647k |
| ‡ These authors contributed to this work equally. |
| § Crystal data for R-1: hexagonal, P63, a = 18.314(1) Å, c = 6.168(1) Å, V = 1791.9(2) Å3, Z = 6, F(000) = 954, ρcalcd = 1.713 g cm−3, μ(MoKα) = 1.579 mm−1 (λ = 0.71073 Å), R1 = 0.045, wR2 = 0.141, GooF = 1.03, Flack parameter = 0.03(3). For S-1: hexagonal, P63, a = 18.288(8) Å, c = 6.168(4) Å, V = 1786.5(1) Å3, Z = 6, F(000) = 954, ρcalcd = 1.718 g cm−3, μ(MoKα) = 1.584 mm−1 (λ = 0.71073 Å), R1 = 0.052, wR2 = 0.123, GooF = 1.05, Flack parameter = 0.06(3). R-2: hexagonal, P63, a = 18.162(1) Å, c = 6.168(7) Å, V = 1762(2) Å3, Z = 6, F(000) = 954, ρcalcd = 1.741 g cm−3, μ(MoKα) = 1.796 mm−1 (λ = 0.71073 Å), R1 = 0.049, wR2 = 0.063, GooF = 0.94, Flack parameter = 0.05(4). For S-2: hexagonal, P63, a = 18.081(2) Å, c = 6.1296(6) Å, V = 1735.5(3) Å3, Z = 6, F(000) = 960, ρcalcd = 1.768 g cm−3, μ(MoKα) = 1.824 mm−1 (λ = 0.71073 Å), R1 = 0.031, wR2 = 0.082, GooF = 1.09, Flack parameter = 0.02(2). CCDC 696828 (R-1), 696829 (S-1), 1060995 (R-2) and 696830 (S-2). |
| This journal is © The Royal Society of Chemistry 2015 |