 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
IrOx core-shell nanocatalysts for cost- and energy-efficient electrochemical water splitting†
Hong Nhan
Nong
a,
Lin
Gan
a,
Elena
Willinger
b,
Detre
Teschner
*b and
Peter
Strasser
*a
aThe Electrochemical Energy, Catalysis and Materials Science Laboratory, Department of Chemistry, Chemical and Materials Engineering Division, Technical University Berlin, Straße des 17. Juni 124, 10623 Berlin, Germany. E-mail: pstrasser@tu-berlin.de
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Department of Inorganic Chemistry, Faradayweg 4-6, D-14195 Berlin, Germany. E-mail: teschner@fhi-berlin.mpg.de
First published on 23rd May 2014
Abstract
A family of dealloyed metal–oxide hybrid (M1M2@M1Ox) core@shell nanoparticle catalysts is demonstrated to provide substantial advances toward more efficient and less expensive electrolytic water splitting. IrNi@IrOx nanoparticles were synthesized from IrNix precursor alloys through selective surface Ni dealloying and controlled surface oxidation of Ir. Detailed depth-resolved insight into chemical structure, composition, morphology, and oxidation state was obtained using spectroscopic, diffraction, and scanning microscopic techniques (XANES, XRD, STEM-EDX, XPS), which confirmed our structural hypotheses at the outset. A 3-fold catalytic activity enhancement for the electrochemical oxygen evolution reaction (OER) over IrO2 and RuO2 benchmark catalysts was observed for the core-shell catalysts on a noble metal mass basis. Also, the active site-based intrinsic turnover frequency (TOF) was greatly enhanced for the most active IrNi@IrOx catalyst. This study documents the successful use of synthetic dealloying for the preparation of metal-oxide hybrid core-shell catalysts. The concept is quite general, can be applied to other noble metal nanoparticles, and points out a path forward to nanostructured proton-exchange-electrolyzer electrodes with dramatically reduced noble metal content.
1. Introduction
Electrocatalytic splitting of water is expected to play a key role in the future sustainable production of hydrogen from electricity. Combined with renewable electric power generation technologies, such as solar electric, solar-thermal, hydro, or wind power plants, it is expected to emerge as a low-emission method for storing excess electricity or for producing abundant quantities of hydrogen fuel as part of a solar refinery.1–6 Electrocatalytic water splitting can be divided into two half redox reactions, hydrogen evolution reaction (HER) on cathode and oxygen evolution reaction (OER) on anode side. The challenge in electrocatalytic water splitting is the anodic multi-electron OER with its extremely sluggish surface kinetics on virtually all known materials. On Pt electrodes, for instance, the OER proceeds about 1010 times more slowly than the HER process.7,8 The OER catalysis is currently a major bottleneck to the development and the application of devices to convert sunlight or electricity into storable fuels.6In principle, the electrochemical OER can be carried out under a wide range of pH conditions.4,9–14 The selection of suitable electrocatalyst materials, however, is critically dependent on the pH of the electrolyte. Acid conditions are demanding in terms of electrode corrosion stability, requiring structurally stable catalysts up to an electrode potential as high as +1.8 V/RHE. This is why acid OER electrocatalysts typically contain noble metal oxides, such as RuOx or IrOx. This makes them expensive and of limited sustainability. Since Beer's early pioneering work,15,16 Ti-supported annealed films of bulk mixed-metal oxide of RuOx, IrOx and TiOx have proven to be the most active and most stable OER electrocatalyst known to date. This family of bulk catalysts came to be known as the “dimensionally stable anode” (DSA). Suitable Ru-Ir mixed metal catalysts show an overpotential of only about 200 mV. Since the early days, it represents the catalyst solution of choice for virtually all commercial acid Polymer Electrolyte Membrane (PEM) electrolyzers.17–20
In order to make RuOx and IrOx-containing OER electrolyzer catalysts more efficient, new materials are sought with (i) lowered overpotential (higher catalytic activity) and (ii) reduced noble metal content.
Rossmeisl et al. reported DFT-computational studies21–25 on OER reactivity trends on well-defined pure metal oxides in terms of oxygen chemisorption energies, yielding a volcano type activity plot, in which Ru oxide and Ir oxide are near the top. From their analysis, oxide materials should exist with more favorable oxygen chemisorption coupled to higher OER reactivity and lower overpotentials than RuO2. However, beyond pure metal oxide catalysts, other known correlations between structure and chemisorption can be invoked and combined with Rossmeisl's concepts to design improved OER catalysts. For instance, it is known that coordinatively unsaturated surface atoms at defects, steps or kinks of surfaces tend to show varied chemisorption compared to terrace atoms and can be leveraged to optimize reactivity.26–35 In addition, tuning the d-band structure of RuOx and IrOx surfaces through non-noble transition metal atoms in underlying atomic layers is expected to be an effective way to enhance catalytic activities.34 This may be based on short-range electronic interaction or based on longer-range geometric strain effects.36
In order to lower the noble metal content (Ru and Ir in case of OER catalysts), mesoporous and nanostructure design of the active phase has attracted much attention.3,37–41 RuO2 nanoparticles (NPs) offer the lowest overpotentials for the OER, yet IrO2 NPs show almost comparable activity and are far more stable3 and are, therefore, of great interest. Still, the extremely scarce nature of Ir renders the Ir NP architecture hardly viable, which is why novel strategies are critically needed to further reduce the amount of Ir in nanoparticle architectures. One strategy involves the well-known use of suitable corrosion-resistant high surface-area supports; another consists of the design of IrM@IrOx hybrid core-shell architectures, where M represents an inexpensive abundant transition metal that helps to tune the intrinsic electrocatalytic activity and lower the noble metal content alike. This is because concentrating Ir in a thin shell of the nanoparticles reduces the required amount of the scarce catalyst significantly. This contribution demonstrates a combination of both strategies.
In the present contribution, we demonstrate the viability and future potential of supported metal-oxide hybrid core-shell OER NP catalysts formed through a dealloying and selective oxidation procedure of bimetallic NPs.42 Exemplified by the IrNi@IrOx system, we compare and contrast their facile preparation to earlier approaches and follow the stepwise emergence of the metal-oxide hybrid core-shell architecture through a combination of High-angle Annular Dark Field Scanning Transmission Electron Microscopy coupled to Energy Dispersive X-ray Spectroscopy (HAADF-STEM-EDX), depth-resolved synchrotron based Photoelectron Spectroscopy (XPS) and X-ray Absorption Spectroscopy (XAS).
The essential idea involves Ir-Ni precursor alloy nanoparticles that were selectively electrochemically dealloyed to form metallic core-shell NPs. Subsequently or simultaneously, they are selectively surface oxidized to form active metal-oxide hybrid core-shell NP catalysts with outstanding catalytic OER activities. Special emphasis is placed on the role of Ni in the alloy material. First, it enhances electrochemical active surface area (ECSA) of the catalysts during selective dealloying. Second, subsurface Ni caused electronic and strain effects resulting in higher intrinsic OER activity, and third, the high Ni core content significantly lowers the total Ir content of the catalyst. To the best of our knowledge, this is the first study on this family of dealloyed metal-oxide hybrid nanoparticle catalysts.
2. Experimental
2.1. Chemicals and materials
Commercially available reagents were used in the synthesis of the Ir and IrNix NPs as received. Nickel acetate tetrahydrate (Ni(ac)2·4H2O) was purchased from Alfa Aesar, and iridium(III) acetate (Ir(ac)3) was purchased from Chempur. Oleic acid, oleylamine, 1,2-tetradecandiol, dichloromethane, dibenzyl ether and ethylene glycol were purchased from Sigma Aldrich. Vulcan XC-72 R carbon black (BET surface area ∼250 m2 g−1) was purchased from Carbot.2.2. Synthesis of Ir and IrNix bimetallic precursor nanoparticle alloys (PA-Ir and PA-IrNix)
2.3. Microscopic and spectroscopy characterization
More details to microscopic and spectroscopy characterization is given in ESI.†
2.4. Electrochemical measurements
All electrochemical measurements were carried out in N2-saturated 0.05 M H2SO4 and repeated on three catalyst films for each catalyst.
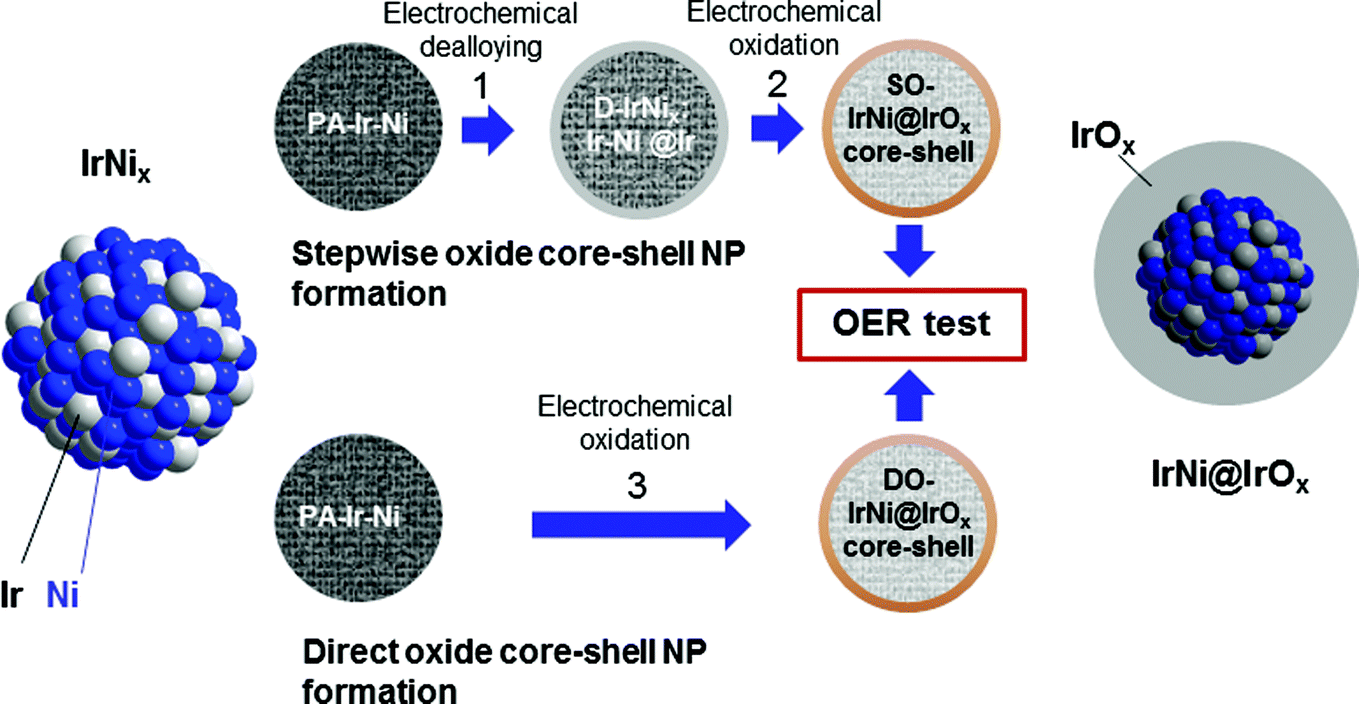 | ||
| Fig. 1 Synthetic protocol for the preparation of SO-IrNi@IrOx and DO-IrNi@IrOx hybrid core-shell nanoparticle catalysts. Precursor IrNi alloys (“PA-IrNi”, and alloy scheme on left, blue: Ni, grey: Ir) are stepwise (SO) or directly (DO) dealloyed and surface oxidized. “D-IrNix” denotes the dealloyed stage. The “SO-IrNix” or “DO-IrNix” nomenclatures emphasize the original stoichiometry of the parent precursor alloy, while the “SO-IrNi@IrOx” and “DO-IrNi@IrOx” nomenclature stresses the chemical structure of the core-shell particles. | ||
3. Results and discussion
3.1. Synthesis and characterization of Ir-Ni precursor alloys (PA-IrNix)
Supported Ir-Ni bimetallic precursor alloy nanoparticles (denoted “PA-IrNix” in Fig. 1) were synthesized with 3 different IrNix ratios (x = 2.3, 3.3, 5.7). Supported pure Ir nanoparticles were used as benchmark catalysts. Composition, morphology and structure of the PA-IrNix NPs were characterized by ICP-OES, powder X-ray diffraction and TEM.The XRD profiles of the IrNix and pure Ir NP catalysts (Fig. 2a) exhibit a peak pattern consistent with a face-centered cubic (fcc) crystal symmetry. There is no superlattice peak discernible below 35° indicating a substitutional solid solution structure throughout. Pure Ir, IrNi2.3 and IrNi3.3 materials exhibited single broad peaks suggesting homogeneous single-phase metallic (alloy) NPs with small crystallite size, on the other hand, IrNi5.7 showed sharper diffraction peaks, indicating larger crystallite size. All diffraction peaks of Ni-containing samples are shifted toward higher 2θ, indicating a contraction of the lattice with the increasing amount of smaller Ni atoms. The relationship between the lattice constant derived from the (111) planes and the composition of the annealed IrNix NPs (Fig. 2b) followed the Vegard's lattice constant-composition law surprisingly well.
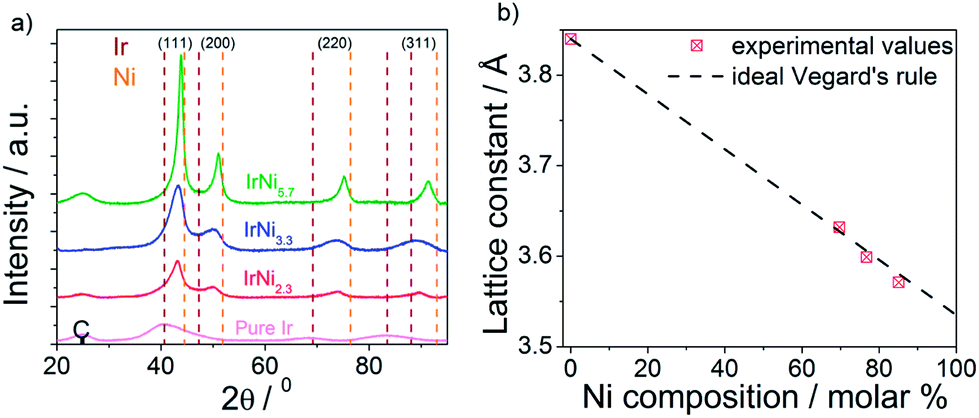 | ||
| Fig. 2 (a) Powder X-ray diffraction patterns of Ir, IrNi2.3, IrNi3.3, IrNi5.7 precursor alloy catalysts; C denotes a support reflection; vertical dashed lines indicate database reflections of pure Ir and pure Ni. (b) the relationship between lattice constant and Ni composition (red symbols) compared to Vegard's law (dashed line). | ||
Bright field TEM images and particle size histograms (Fig. 3) showed narrow particle size distribution of the pure Ir and IrNix bimetallic NPs up to the IrNi3.3 ratio. The mean particle size increased with increasing bulk Ni content. Composition, crystallite size and particle size of Ir and IrNix nanoparticles are tabulated in Table S1.†
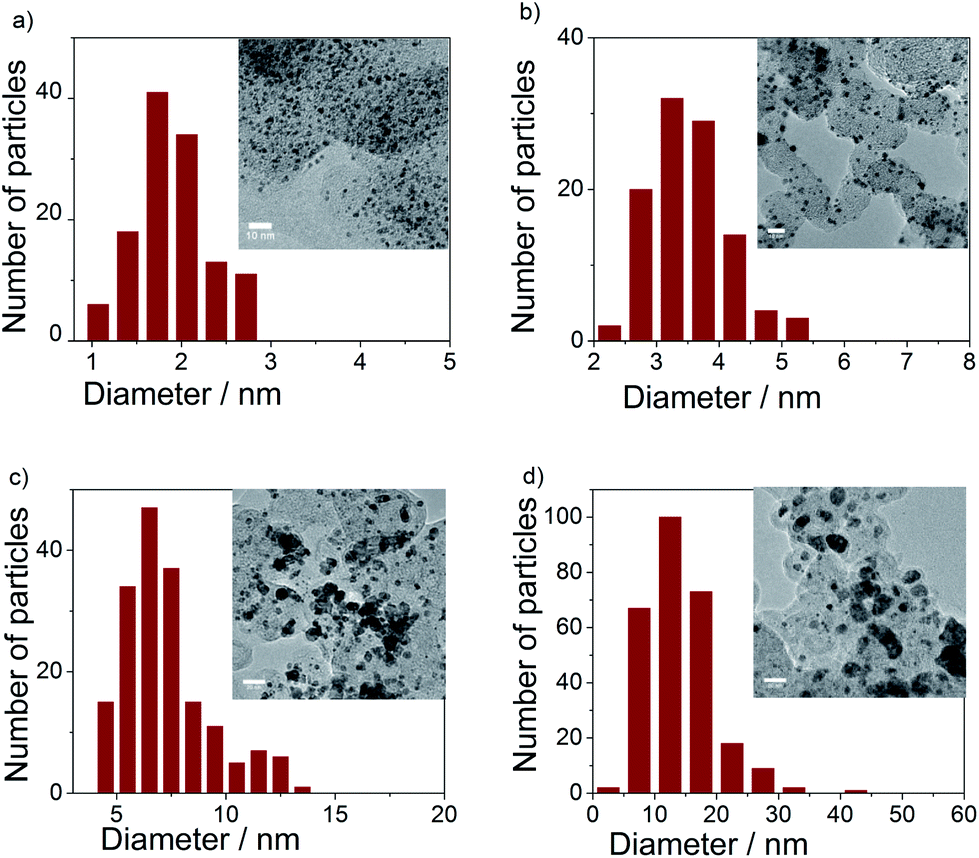 | ||
| Fig. 3 TEM images and particle size histograms of (a) Ir, (b) IrNi2.3, (c) IrNi3.3, and (d) IrNi5.7 precursor alloys. | ||
3.2. Preparation of IrNi@IrOx hybrid core-shell nanoparticles
Comparative inspection of the cyclic voltammograms (CVs) of the pristine PA-IrNix and dealloyed D-IrNix catalysts (Fig. 4a) showed the typical hydrogen surface electrochemistry of Ir manifested by underpotential hydrogen deposition (Hupd) and stripping peaks between +0.05 and +0.4 V. Unlike pure Ir, however, the first scans of the PA-IrNix catalysts (dashed lines) displayed an oxidative wave around +0.3 V (best seen for the Ni rich catalysts) evidencing the electrochemical dissolution of surface Ni atoms.36 In the CV profiles of D-IrNix NPs (solid lines), however, these Ni oxidation features became absent, restoring the typical pure Ir voltammetric shape; we can conclude that, at this point, the dissolution of surface Ni atoms has yielded a stable metallic IrNi@Ir core-shell structure, which will be backed up below by spectroscopy.
 | ||
| Fig. 4 (a) Voltammetric profiles of the precursor alloy PA-IrNix NPs (dashed) and dealloyed D-IrNix NPs (solid) in N2-saturated 0.05 M H2SO4 aqueous solution at 20 mV s−1. (b) Comparison of the electrochemically active surface areas of PA-IrNix and D-IrNix NPs. | ||
The initial electrochemical surface areas of the PA-IrNix NPs dropped with increasing Ni content (Fig. 4b), which can be explained by increasing particle size and more Ni enriched particle surfaces. In contrast, D-IrNi3.3 and D-IrNi5.7 NPs showed a 3 and 7 fold enhancement in the ECSA in comparison to their respective pristine state. The trend in ECSA of the dealloyed NPs with increasing Ni content is fully plausible considering that roughness and defect densities increase with more initial surface Ni content.36
In order to confirm our hypothesis about the formation of a metallic core-shell structure, and to get further atomic-scale insight into the nanostructure of the catalysts before and after the dealloying, we performed high-angle annular dark-field imaging (HAADF)-STEM and STEM-EDX line scans of the IrNi3.3 material. Fig. S1a† shows dark field images of well-alloyed PA-IrNi3.3 catalyst particles with largely homogeneous chemical composition. On the other hand, dealloyed D-IrNi3.3 NPs (Fig. S1b†) exhibited bright Z-contrast shells surrounding darker inner parts (cores), evidencing the formation of IrNi@Ir core shell nanostructures. STEM-EDX line scans of PA-IrNi3.3 and D-IrNi3.3 NPs (Fig. 5) corroborate these conclusions unambiguously.36,47 Upon dealloying, the homogeneous Ir, Ni distribution yields to a core shell type line scan of D-IrNi3.3 with two symmetric Ir shell peaks. Interestingly, the morphology of the dealloyed nanoparticles strongly depended on particle size, while smaller NPs favor Ni-richer core-shell structures, larger NPs appear to undergo severe Ni leaching in their transition to core-shell structures.
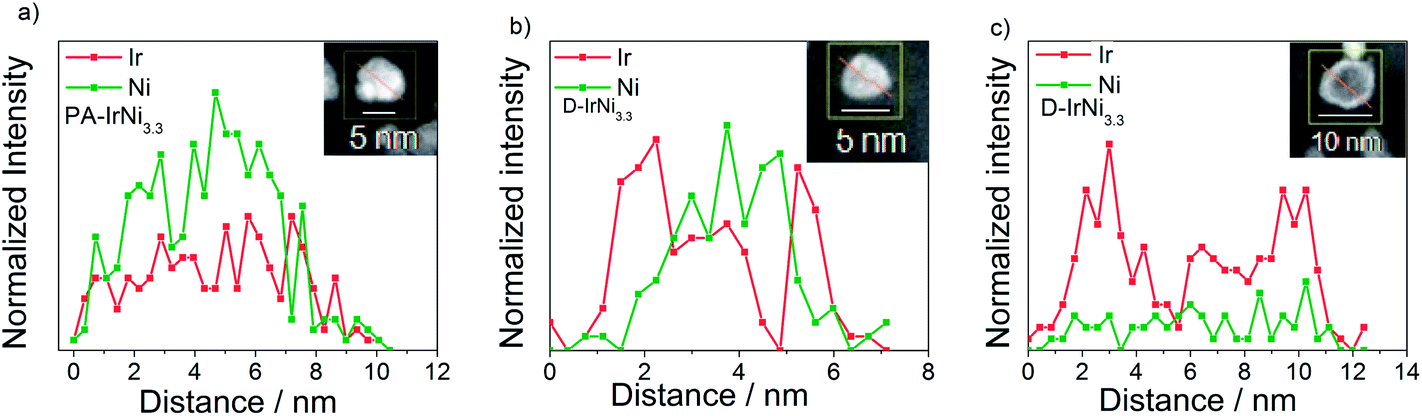 | ||
| Fig. 5 STEM-EDX line profiles of Ir and Ni in (a) PA-IrNi3.3 and (b and c) D-IrNi3.3. The red arrows in the corresponding NPs (see insets) indicate the directions of the EDX line profiles. | ||
To corroborate our core-shell structure hypothesis further for the metallic IrNi@Ir materials, we performed depth profiled X-ray Photoelectron Spectroscopy (XPS) at two different kinetic photoelectron energies (KE) as well as X-ray Absorption Near Edge Structure (XANES). Survey XPS spectra (Fig. S2†) confirmed the absence of impurities in the precursor alloy or any of the core-shell materials. The chemical state of the elements of interest (Ni and Ir) was examined in high resolution and depth resolved. The Ni 2p region of PA-IrNi3.3 (Fig. 6a, red lines, solid: 210 eV KE, dotted: 550 eV KE) indicated the prevalence of oxidic Ni species highly reminiscent of NiO.48 This is plausible given the high Ni content of the pristine NPs.
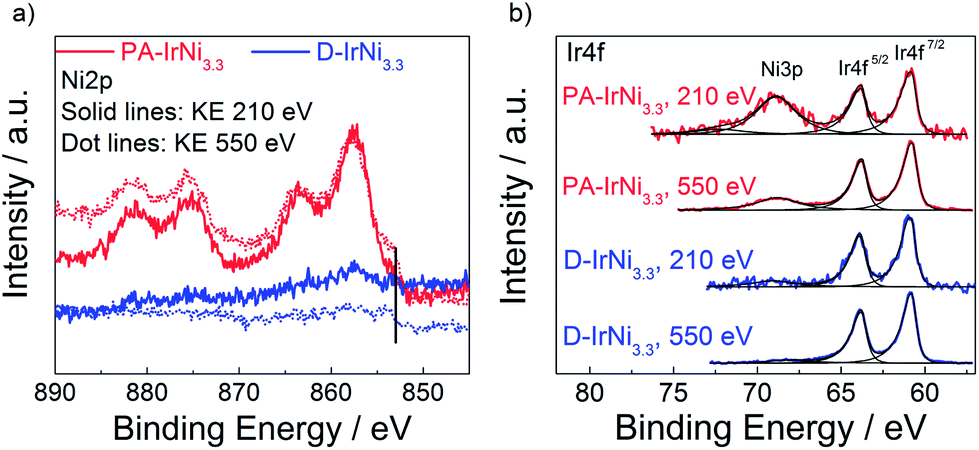 | ||
| Fig. 6 (a) Ni2p and (b) Ir4f XPS spectra of PA-IrNi3.3 and D-IrNi3.3 at photoelectron kinetic energy (KE) of 210 eV and 550 eV. Vertical line in (a) marks the core level shift of metallic Ni. | ||
A low binding energy shoulder around 853 eV marked in Fig. 6a indicates a minor metallic Ni contribution to the overall line shape. Depth profiling of Ni2p with two different kinetic energies (210 eV and 550 eV) clearly evidenced the metallic component of Ni being more abundant when probing deeper into the NPs. This is consistent with the notion that the core of the particle consists of metallic Ni, while the surface underwent oxidation. Song et al.49 claimed the formation of ∼1 nm oxide layer on Ni NPs occurred during the production or storage period, which might be caused by the non-uniform distribution of defects of nickel crystal structures, i.e. areas with large defects could result in high oxidation levels and vice versa. Similarly, in our case, Ni in the near-surface region of PA-IrNi3.3 NPs was oxidized, probably in an amorphous XRD invisible state.
Upon dealloying, we recorded dramatically reduced NiOx intensities (blue solid, 210 eV KE, and dotted lines, 550 eV KE), a nearly six-fold reduction in the estimated Ni/Ir molar ratio (see Table 1), and a sustained contribution of metallic Ni0 in the subsurface region. All these findings are in excellent agreement with expectations and microscopic data and strengthened our hypothesis as to the electrochemical formation of a metallic IrNi@Ir core-shell catalyst structure.
| Sample | Ni/Ir ratio at 550 eV KE | O/(Ir + Ni) ratio at 550 eV KE |
|---|---|---|
| PA-IrNi3.3 | 6.1 | 1.6 |
| D-IrNi3.3 | 0.93 | 3.3 |
| SO-IrNi3.3 | 0.09 | 5.2 |
| DO-IrNi3.3 | 0.14 | 4.1 |
| DO-IrNi3.3-Stabil | 0.045 | 6.0 |
XANES Ni L edge spectra of PA-IrNi3.3 and D-IrNi3.3 (Fig. S3a†) revealed slight but characteristic differences, suggesting more NiO contribution (∼855 eV) in the PA-IrNi3.3 catalyst, whereas more metallic Ni0 contributions in the D-IrNi3.3 material (satellite structure in Fig. S3a and S3b† at 859 eV) in line with the XPS results.
The XPS Ir4f region of the PA-IrNi3.3 and D-IrNi3.3 catalysts (see Fig. 6b) indicated the prevalence of metallic Ir0 (BE ∼60.8 eV). Interestingly, the Ir4f region overlapped with the Ni 3p region. This is why Ni-related 3p peaks appear. These peaks corroborated our conclusions from the 2p region: Ni3p of the precursor alloy evidenced a NiOx-covered surface. After dealloying, Ir remained in fully metallic state; however the intensity of Ni3p significantly decreased, which is commensurate with the selective removal of Ni from the surface. Detailed quantitative evaluations using the XPS Ir4f, Ni2p and O1s core levels at 550 eV KE in ultra high vacuum (UHV) are provided in Table 1. The increase in the O/(Ir + Ni) ratio upon dealloying (PA to D) despite the dissolution of NiOx species is explained by the surface oxidation of the carbon support after potential cycling.
Fig. S4a† displays the evolution of the surface chemistry of the D-IrNi3.3 during voltammetric surface oxidation. The formation of irreversible Ir oxide is clearly evidenced by the diminishing hydrogen adsorption (+0.05 V to +0.4 V) with a strongly emerging Ir(III)/Ir(IV) redox wave (+0.8 V to +1.2 V). The characteristic Ir(III)/Ir(IV) redox couple (see Fig. S4b†) was used to estimate the molar amount of catalytically active Ir atoms for each catalyst as given in Table S2.50,51† While we are aware that the values in Table S2† likely represent an upper bound of the active OER sites, we used them to calculate intrinsic turn-over-frequency (TOF) type kinetic rates, equivalent to lower bounds of the OER activity. DO-IrNix samples showed larger numbers of active Ir sites than SO-IrNix samples, presumably due to increased surface roughness and defects during the massive surface atomic rearrangements during simultaneous dealloying and oxidation.
To directly confirm the core shell structure of the SO-IrNi@IrOx and DO-IrNi@IrOx catalysts synchrotron-based depth-resolved XPS and XANES studies were performed. Flat spectra at both information depths in the XPS Ni2p region of both SO-IrNi3.3 and DO-IrNi3.3 (Fig. S5,† green and orange lines) clearly proved an essentially complete removal of near surface Ni. Quantitative metal ratios are presented in Table 1. Similarly, Ni L Edge XANES spectra of both SO-IrNi3.3 and DO-IrNi3.3 (Fig. S3a†) resembled those of the NiO standards.
Depth resolved Ir4f core level spectra of the SO-IrNi3.3 and DO-IrNi3.3 catalysts (Fig. 7) were clearly shifted to higher binding energy, consistent with the formation of surface IrOx. The Ir XPS spectra at low KE 210 eV indicated that Ir atoms in the surface layer were completely oxidized, however, depth profiling at higher KE (550 and 1200 eV) also showed that Ir metallic components were present at larger information depth. This corroborates our structural hypothesis that the NP cores of the catalysts still contain metallic alloy. It should be noted here that the shape of the Ir4f peak lacks the characteristic asymmetry of IrO2,52 suggesting that the Ir oxide component here cannot be IrO2 itself, but rather a mixture of ill-defined oxide-hydroxide, with potentially numerous defects and thus varying oxidation states.53 Concerning the thickness and stability of the formed Ir oxide layer, as well as the real oxidation states of Ir during oxidation step and oxygen evolution reaction, further investigation is necessary.
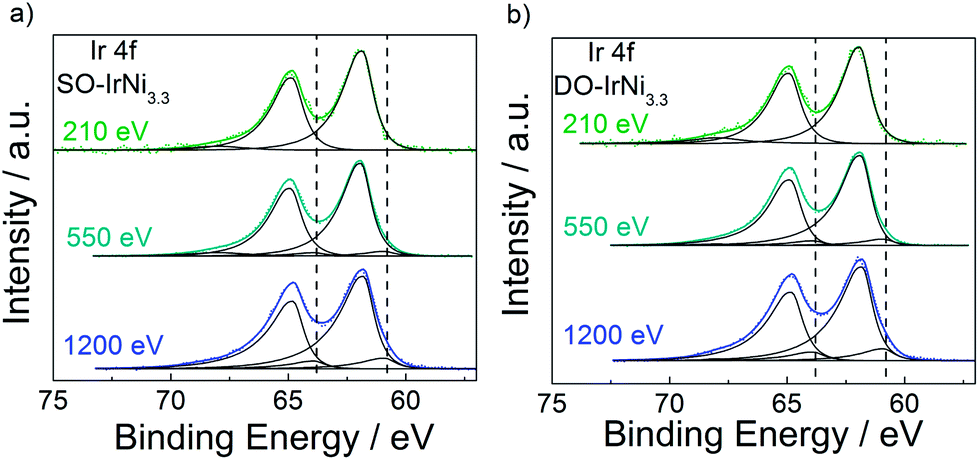 | ||
| Fig. 7 Ir4f core level spectrum of (a) SO-IrNi3.3 and (b) DO-IrNi3.3 at photoelectron kinetic energy of 210, 550 and 1200 eV. Dash lines indicate peak position of metallic Ir as found in PA-IrNi3.3. | ||
All in all, the extensive and comprehensive HAADF-STEM, STEM-EDX, XANES, and XPS results evidence the successful synthesis of metallic and metal-oxide hybrid core-shell nanoparticle by selective removal of Ni and surface oxidation, which confirms our structural hypotheses illustrated in Fig. 1.
3.3. Electrochemical OER activity of IrNi@IrOx core-shell nanoparticles
In order to correlate the metal-oxide hybrid core-shell structure with surface catalytic activity for the oxygen evolution reaction (OER) under approximate conditions of membrane electrolyzers, the SO-IrNi@IrOx and DO-IrNi@IrOx core-shell catalysts were subjected to a sweep voltammetry protocol in highly acidic electrolyte environment. The electrochemical activities were corrected for the uncompensated ohmic resistance of the set-up and electrolyte (IR correction) and for the capacitive current contribution. Finally the faradaic currents were normalized by the Ir mass (mass specific activity) or by the molar amount of the accessible Ir redox centers (intrinsic activity). The sweep voltammetry of all catalyst along with their Ir-mass-based and specific activities at 0.25 V overpotential are presented in Fig. 8.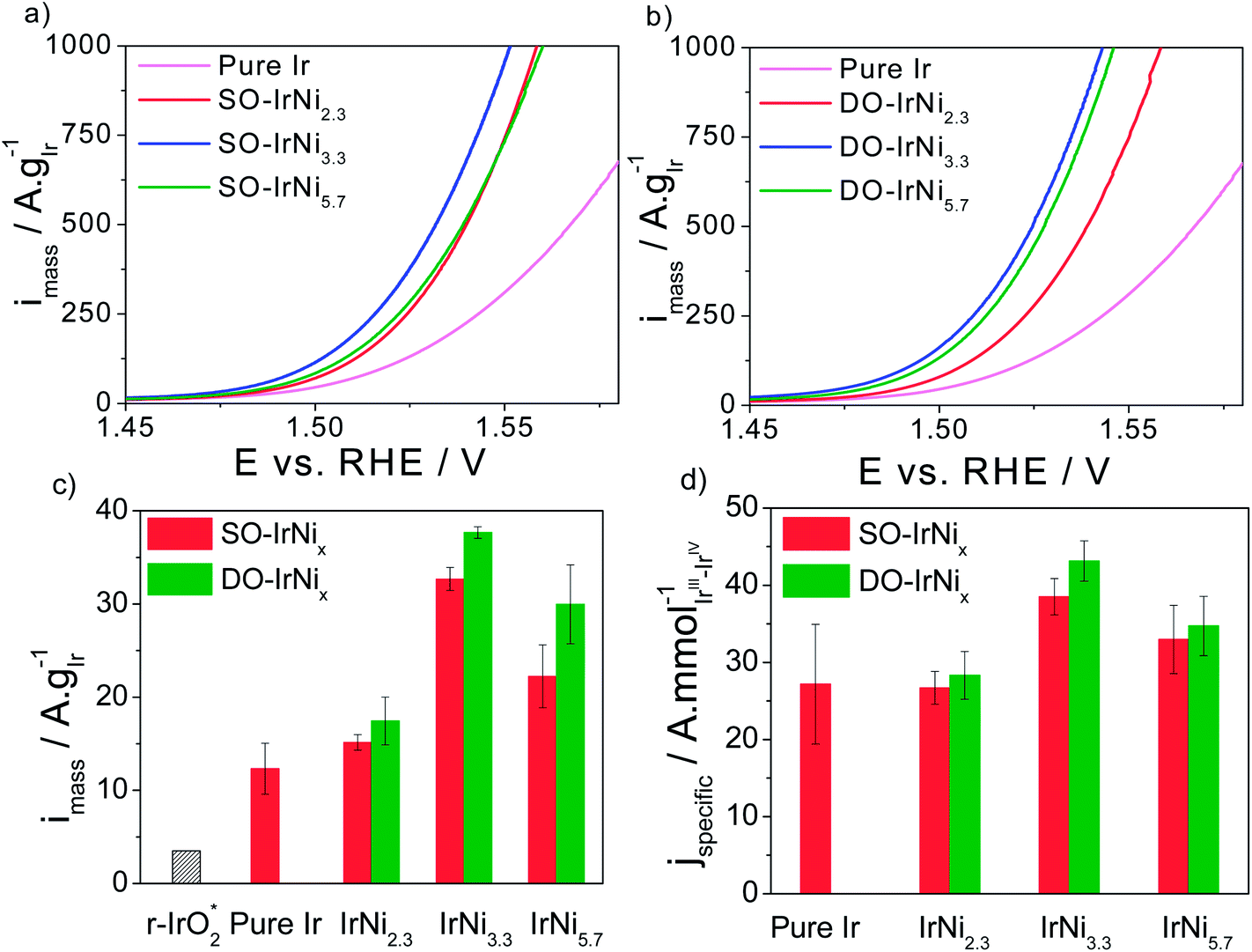 | ||
| Fig. 8 (a and b) Rotating Disk Electrode (RDE) sweep voltammetry and catalytic oxygen evolution reaction (OER) activities of stepwise oxidized (SO) IrNix and directly oxidized (DO) IrNix core shell nanoparticle catalysts, in comparison to pure Ir nanoparticles. (c) Ir mass based activities and (d) specific activities at 0.25 V overpotential. Conditions: 0.05 M H2SO4, 1600 rpm, scan rate 5 mV s−1. “r-IrO2” denotes the Ir-mass based activity of IrO2 rutile NPs at 0.25 V overpotential.37 | ||
Quite obviously, all Ni-containing nanoparticle catalysts are significantly more active for the OER on a mass and active site-specific basis than pure Ir. The most active catalysts are the SO-IrNi3.3 and the DO-IrNi3.3 showing a 3 times higher mass activity compared to pure Ir. This implies a three times higher electrolyzer hydrogen output at constant input voltage. Directly oxidized core shell catalysts showed even slightly higher OER mass activity than that of stepwise oxidized samples, likely due to higher surface roughness and more catalytic sites.
In comparison to the open literature, our IrNi@IrOx metal-oxide core shell catalysts nanoparticles showed substantially higher mass activity, even compared to a rutile iridium oxide particle catalyst (“r-IrO2” in Fig. 8), which is often taken as a benchmark Ir OER catalyst.37 In particular, the DO-IrNi3.3 core shell catalyst exhibited a 10 times higher mass activity than r-IrO2. Moreover, although ruthenium has been reported to be the most active catalyst for OER in acidic media, all of our Ni-containing catalysts are more active for OER than a rutile r-RuO2 nanoparticle catalyst (Ru mass activity of 11 A g−1RuO2 or 14.5 A g−1Ru compared to 30–40 A g−1Ir for DO-IrNi3.3, an almost 3 times activity enhancement).37 At higher overpotentials, Wei Hu et al.54 reported a noble metal mass activity of three-dimensional ordered macroporous IrO2 of 500 A g−1IrO2 (or 583 A g−1Ir) at 1.60 V vs. RHE; for comparison, our DO-IrNi3.3 reaches the same value already at 1.530 V vs. RHE corroborating the mass efficient OER catalysis.
In terms of Ir-site based specific activities, IrNi2.3 sample shows similar activity to pure Ir, while the two Ni-rich samples (IrNi3.3 and IrNi5.7) showed substantially higher specific activity than Ir, however the enhancement remain below the mass-based value. This is explained by the fact that the number of active sites is an upper bound, rendering the specific activities a lower bound of actual OER rate.
In order to test stability of the nanocatalysts, chronopotentiometric measurements were carried out at room temperature in aqueous 0.05 M H2SO4 electrolyte for the pure Ir and DO-IrNi3.3 nanoparticles (Fig. 9). The potential increased slightly during the first 30 minutes and then remained constant throughout the tests, indicating stable behavior of the nanocatalysts.
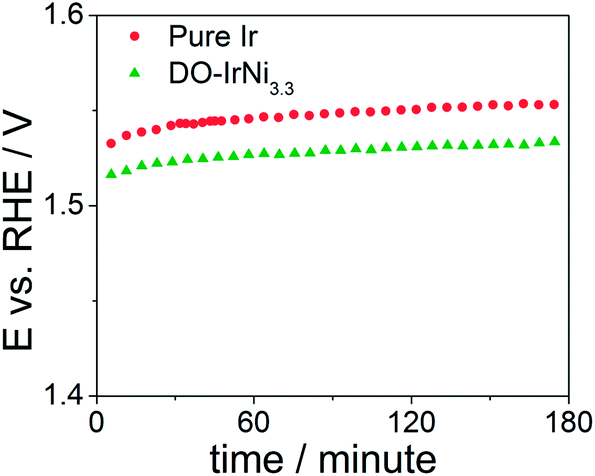 | ||
| Fig. 9 Chronopotentiometric measurements of pure Ir and directly oxidized (DO) IrNi3.3 core-shell nanoparticles at a current density of 1 mA cm−2. | ||
XPS Ir4f spectra at KE 550 eV of directly oxidized IrNi3.3 before (denoted as DO-IrNi3.3) and after stability test (denoted as DO-IrNi3.3-Stabil) were compared (Fig. S6†). The Ir4f spectrum shapes were quite similar, though the fits suggest that a small metallic contribution of the DO-IrNi3.3 sample decreased further during the stability test. This indicates a prolonged oxidation of the particles from the outside in to even larger depths. Composition evaluation of DO-IrNi3.3-Stabil (Table 1) gave an Ir/Ni ratio of 0.045, showing that some small Ni was further removed from the surface region during stability test.
4. Conclusions
We have presented a family of dealloyed metal-oxide hybrid (M1M2@M1Ox) core@shell nanoparticle catalysts and have demonstrated, exemplified by the IrNi@IrOx system, that this structural and compositional class of nanocatalysts offers substantial advances in terms of more efficient and less expensive electrolytic water splitting for acid electrolyzer applications.Using extensive microscopic and spectroscopic analysis, largely with depth resolution, we followed the evolution of the IrNi@IrOx metal oxide core shell particles step-by-step from their IrNi bimetallic precursor stage, through the metallic core-shell state to the final oxide shell/alloy core architecture. The final catalysts have a nanometer scale almost pure IrOx surface, while the inner core region became increasingly metallic and richer in Ni.
Correlating the structural characteristic with OER activity we concluded that the core-shell catalysts perform substantially more active (DO-IrNi3.3 showed about 3× higher compared to electrochemically oxidized Ir and rutile RuO2, and 10× higher compared to rutile IrO2) and thus thermodynamically more efficient on a noble metal mass and an active site basis during the electrocatalytic oxygen evolution.
The dealloyed metal-oxide hybrid (M1M2@M1Ox) core-shell catalyst concept represents a quite general strategy to lower the noble-metal content of nanoparticle oxide catalysts for other technologies or applications, including but not limited to energy storage and conversion such as electrolyzers, supercapacitors, or metal-air batteries. Deploying the current catalysts in polymer electrolyte membrane electrolyzer anodes, for instance, offers the prospect of lowering the required Ir content for a given hydrogen production rate by a full factor of 3.
Acknowledgements
We thank Dr. M. G. Willinger for collaboration regarding the STEM-EDX data collection. We thank the Zentraleinrichtung für Elektronenmikroskopie (Zelmi) of the Technical University Berlin for their support with TEM technique. Financial support by the German Research Foundation (DFG) through grant STR 596/3-1 under the Priority Program 1613 “Regeneratively formed fuels by light driven water splitting” is gratefully acknowledged. Hong Nhan Nong acknowledges financial support by the Ministry of Education and Training of The Socialist Republic of Vietnam (MOET) and the German Academic Exchange Service (DAAD). This project received partial funding from the Federal Ministry of Education and Research under the grant reference number 03SF0433A “MEOKATS”.References
-
Chemical Energy Storage, ed. R. Schloegl, De Gruyter, Berlin, 2013 Search PubMed
.
- R. Forgie, G. Bugosh, K. C. Neyerlin, Z. C. Liu and P. Strasser, Electrochem. Solid-State Lett., 2010, 13, D36 CrossRef PubMed
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765 CrossRef CAS
- A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Energy Environ. Sci., 2013, 6, 2745 CAS
- K. C. Neyerlin, G. Bugosh, R. Forgie, Z. Liu and P. J. Strasser, Electrochem. Soc., 2009, 156, B363 CrossRef CAS PubMed
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS
-
J. P. Hoare, The Electrochemistry of Oxygen, Wiley, New York, 1968 Search PubMed
-
R. C. O'Hayre, S.-W. Cha, W. Colella and F. B. Prinz, Fuel Cell Fundamentals, Wiley, New York, 2006 Search PubMed
- T. C. Wen and C. C. Hu, J. Electrochem. Soc., 1992, 139, 2158 CrossRef CAS PubMed
- V. V. Kuznetsov, S. A. Chepeleva, M. M. Gol'din, V. N. Kudryavtsev, V. N. Fateev and A. G. Volkov, Russ. J. Electrochem., 2005, 41, 933 CrossRef CAS
- H. Ma, C. Liu, J. Liao, Y. Su, X. Xue and W. Xing, J. Mol. Catal. A: Chem., 2006, 247, 7 CrossRef CAS PubMed
- A. Marshall, B. Børresen, G. Hagen, S. Sunde, M. Tsypkin and R. Tunold, Russ. J. Electrochem., 2006, 42, 1134 CrossRef CAS
- A. Marshall, B. Børresen, G. Hagen, M. Tsypkin and R. Tunold, Electrochim. Acta, 2006, 51, 3161 CrossRef CAS PubMed
- V. V. Panić, A. B. Dekanski, S. K. Milonjić, V. B. Mišković-Stanković and B. Ž. Nikolić, Russ. J. Electrochem., 2006, 42, 1055 CrossRef
-
H. B. Beer, Electrode and coating thereof, US patent, 3632498, 1972 Search PubMed
-
H. B. Beer, Electrode having platinum metal oxide coating thereon, and method of use thereof, US patent, 3711385, 1973 Search PubMed
- L. Ma, S. Sui and Y. Zhai, Int. J. Hydrogen Energy, 2009, 34, 678 CrossRef CAS PubMed
- P. Millet, R. Ngameni, S. A. Grigoriev, N. Mbemba, F. Brisset, A. Ranjbari and C. Etievant, Int. J. Hydrogen Energy, 2010, 35, 5043 CrossRef CAS PubMed
- P. Millet, N. Mbemba, S. A. Grigoriev, V. N. Fateev, A. Aukauloo and C. Etiévant, Int. J. Hydrogen Energy, 2011, 36, 4134 CrossRef CAS PubMed
- S. Song, H. Zhang, X. Ma, Z. Shao, R. T. Baker and B. Yi, Int. J. Hydrogen Energy, 2008, 33, 4955 CrossRef CAS PubMed
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37 CrossRef CAS PubMed
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159 CrossRef CAS
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83 CrossRef CAS PubMed
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178 CrossRef CAS PubMed
- J. Rossmeisl, K. Dimitrievski, P. Siegbahn and J. K. Nørskov, J. Phys. Chem. C, 2007, 111, 18821 CAS
- J. Greeley, J. K. Norskov and M. Mavrikakis, Annu. Rev. Phys. Chem., 2002, 53, 319 CrossRef CAS PubMed
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed
- P. Strasser, S. Koh and J. Greeley, Phys. Chem. Chem. Phys., 2008, 10, 3670 RSC
- P. Ferrin, A. U. Nilekar, J. Greeley, M. Mavrikakis and J. Rossmeisl, Surf. Sci., 2008, 602, 3424 CrossRef CAS PubMed
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897 CrossRef CAS PubMed
- J. Greeley, J. K. Nørskov, L. A. Kibler, A. M. El-Aziz and D. M. Kolb, ChemPhysChem, 2006, 7, 1032 CrossRef CAS PubMed
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909 CrossRef CAS PubMed
- M. P. Andersson, T. Bligaard, A. Kustov, K. E. Larsen, J. Greeley, T. Johannessen, C. H. Christensen and J. K. Nørskov, J. Catal., 2006, 239, 501 CrossRef CAS PubMed
- J. Greeley and J. K. Nørskov, Surf. Sci., 2005, 592, 104 CrossRef CAS PubMed
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810 CrossRef CAS PubMed
- L. Gan, M. Heggen, S. Rudi and P. Strasser, Nano Lett., 2012, 12, 5423 CrossRef CAS PubMed
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. J. Shao-Horn, Phys. Chem. Lett, 2012, 3, 399 CrossRef CAS
- T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958 CAS
- Y. X. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. J. Mallouk, Phys. Chem. Lett, 2011, 2, 402 CrossRef CAS
- A. Marshall and R. Haverkamp, J. Mater. Sci., 2012, 47, 1135 CrossRef CAS
- E. Ortel, T. Reier, P. Strasser and R. Kraehnert, Chem. Mater., 2011, 23, 3201 CrossRef CAS
- M. Oezaslan, F. Hasche and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 3273 CrossRef CAS
- K. Ahrenstorf, O. Albrecht, H. Heller, A. Kornowski, D. Görlitz and H. Weller, Small, 2007, 3, 271 CrossRef CAS PubMed
- V. A. Ribeiro, O. V. Correa, A. O. Neto, M. Linardi and E. V. Spinacé, Appl. Catal., A, 2010, 372, 162 CrossRef CAS PubMed
- S. Rudi, X. Tuaev and P. Strasser, Electrocatalysis, 2012, 3, 265 CrossRef CAS PubMed
- R. Woods, J. Electroanal. Chem. Interfacial Electrochem., 1974, 49, 217 CrossRef CAS
- C. Cui, L. Gan, H.-H. Li, S.-H. Yu, M. Heggen and P. Strasser, Nano Lett., 2012, 12, 5885 CrossRef CAS PubMed
- D. Alders, F. C. Voogt, T. Hibma and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 7716 CrossRef CAS
- P. Song, D. Wen, Z. X. Guo and T. Korakianitis, Phys. Chem. Chem. Phys., 2008, 10, 5057 RSC
- C. P. De Pauli and S. Trasatti, J. Electroanal. Chem., 1995, 396, 161 CrossRef
- T. A. F. Lassali, J. F. C. Boodts and L. O. S. Bulhões, J. Appl. Electrochem., 2000, 30, 625 CrossRef CAS
- G. K. Wertheim and H. J. Guggenheim, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 4680 CrossRef CAS
- J. Juodkazyte, B. Sebeka, I. Valsiunas and K. Juodkazis, Electroanalysis, 2005, 17, 947 CrossRef CAS
- W. Hu, Y. Wang, X. Hu, Y. Zhou and S. Chen, J. Mater. Chem., 2012, 22, 6010 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc01065e |
| This journal is © The Royal Society of Chemistry 2014 |