 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation†
Yimeng
Ma
,
Stephanie R.
Pendlebury
,
Anna
Reynal
,
Florian
Le Formal
and
James R.
Durrant
*
Department of Chemistry, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: j.durrant@imperial.ac.uk
First published on 8th May 2014
Abstract
The dynamics of photogenerated holes in undoped BiVO4 photoanodes for water splitting were studied using transient absorption spectroscopy, correlated with photoelectrochemical and transient photocurrent data. Transient absorption signals of photogenerated holes were identified using electron/hole scavengers and applied electrical bias in a complete photoelectrochemical cell. The yield of long-lived (0.1–1 s) photogenerated holes is observed to correlate as a function of applied electrical bias with the width of the space charge layer, as determined by electrochemical impedance spectroscopy. The transient absorption decay time constant of these long-lived holes is also observed to be dependent upon the applied bias, assigned to kinetic competition between water oxidation and recombination of these surface accumulated holes with bulk electrons across the space charge layer. The time constant for this slow recombination measured with transient absorption spectroscopy is shown to match the time constant of back electron transfer from the external circuit determined from chopped light transient photocurrent measurements, thus providing strong evidence for these assignments. The yield of water oxidation determined from these measurements, including consideration of both the yield of long-lived holes, and the fraction of these holes which are lost due to back electron/hole recombination, is observed to be in good agreement with the photocurrent density measured for BiVO4 photoanodes as a function of bias under continuous irradiation. Overall these results indicate two distinct recombination processes which limit photocurrent generation in BiVO4 photoanodes: firstly rapid (≤microseconds) electron/hole recombination, and secondly recombination of surface-accumulated holes with bulk BiVO4 electrons. This second ‘back electron transfer’ recombination occurs on the milliseconds–seconds timescale, and is only avoided at strong anodic biases where the potential drop across the space charge layer provides a sufficiently large energetic barrier to prevent this recombination process.
Introduction
Solar water splitting using inorganic semiconductors has the potential to produce clean and renewable molecular fuels.1 Among the semiconductors investigated for light-driven water oxidation, n-type metal oxides with small optical band gaps are of particular interest due to their potentially low fabrication costs and visible light absorption. Bismuth vanadate (BiVO4) has attracted particular interest in this regard. Its band gap of 2.4–2.5 eV (absorption edge: 500–520 nm) indicates that it could in theory produce ∼6.2–7.5 mA cm−2 photocurrent, whilst its valence band edge (∼2.5 VRHE) provides a strong driving force for water oxidation by photogenerated holes. Whilst initial reported current densities were relatively modest, various strategies have been investigated to increase the efficiency of BiVO4 photoanodes,2 including doping,3–6 heterojunctions,7–9 crystal facet studies,10,11 and surface treatments.6,12–16 For example, van de Krol and co-workers have recently reported a promising photocurrent of 3.6 mA cm−2 at 1.23 VRHE (potential relative to the reversible hydrogen electrode) using a gradient tungsten-doped BiVO4 photoanode covered with cobalt phosphate (CoPi).17 However, for BiVO4 photoanodes without catalysts, such high photocurrent densities are only observed in the presence of significant external bias, with an onset potential for photocurrent generation typically ∼0.6 VRHE and saturated photocurrent only achieved for voltages >1.3 VRHE. These voltages are substantially anodic of the flat band potential of BiVO4 conduction band, ∼0.1 VRHE, and limit the overall thermodynamic efficiencies of BiVO4 photoanodes for solar energy conversion. The requirement for such anodic voltages has been related to the requirement of forming a space charge layer at the semiconductor/electrolyte interface in order to reduce charge recombination in the bulk and at the semiconductor/electrolyte interface, thought to be major loss processes in these electrodes.16 It is the bias dependence of these recombination processes, and their competition with water oxidation by photogenerated BiVO4 holes which is the focus of the study reported herein.Functional studies of recombination and water oxidation kinetics in BiVO4 have to date largely been limited to photoelectrochemical analyses,3,13,16 with for example some studies indicating that that ‘surface recombination’ in BiVO4 photoanodes limits solar conversion efficiencies.13,16 Surface recombination losses have also been related to current spikes observed under chopped irradiation at potentials near the onset of steady state photocurrent generation.13,18,19 We have recently shown that transient absorption spectroscopy is a complementary tool for studying the kinetic competition between charge recombination and water oxidation in metal oxide photoanodes.20–23 Such transient optical studies, whilst being a pulsed technique and therefore sometimes more difficult to relate to steady state operation, allow the direct observation of the dynamics of the photoinduced holes responsible for water oxidation. This contrasts with photoelectrochemical and impedance analyses of such n-type photoelectrodes, where hole dynamics are inferred from electron currents monitored via the external circuit. Transient absorption spectroscopy has previously been used to investigate the dynamics of photogenerated charge carriers in TiO2,21,24,25 WO3,26 and α-Fe2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20,27 photoanodes on timescales ranging from femtoseconds to seconds. Transient absorption spectroscopic studies of BiVO4 photoelectrodes have been very limited to date,22 and have not addressed the kinetic competition between charge recombination and water oxidation, nor the impact of this competition upon photoanode efficiency. A detailed investigation of charge carrier dynamics in α-Fe2O3 photoelectrodes has found that applied anodic bias can retard charge recombination losses and result in the generation of long-lived (lifetimes of 100 ms–seconds) holes, and correlated these data with photocurrent generation.20,23 Analogous data have also been observed for TiO2 photoanodes.21,24
20,27 photoanodes on timescales ranging from femtoseconds to seconds. Transient absorption spectroscopic studies of BiVO4 photoelectrodes have been very limited to date,22 and have not addressed the kinetic competition between charge recombination and water oxidation, nor the impact of this competition upon photoanode efficiency. A detailed investigation of charge carrier dynamics in α-Fe2O3 photoelectrodes has found that applied anodic bias can retard charge recombination losses and result in the generation of long-lived (lifetimes of 100 ms–seconds) holes, and correlated these data with photocurrent generation.20,23 Analogous data have also been observed for TiO2 photoanodes.21,24
Herein, we report the first transient absorption study of water oxidation on undoped BiVO4 photoanodes on microsecond to second timescales – the timescales of most relevance to water oxidation. BiVO4 photoanodes were fabricated using modified metal–organic deposition (MOD), according to the method developed by Sayama et al.28 Long-lived transient absorption signals observed following low intensity pulsed excitation are assigned to the absorption of photogenerated BiVO4 holes by the use of electron and hole scavengers, and applied electrical bias. In the photoelectrochemical cell, the dynamics of photogenerated holes in BiVO4 are studied as a function of applied bias and laser excitation intensity. These hole dynamics are correlated with the width of the space charge layer calculated from impedance analysis. Measurements of chopped light transient photocurrents are employed to study the kinetics of recombination of bulk BiVO4 electrons with long-lived surface holes. The impact of the relative rates of recombination and water oxidation upon the efficiency of water photo-oxidation are discussed.
Experimental
Fabrication of BiVO4 photoanodes
Chemicals were purchased from Sigma-Aldrich, unless otherwise stated, in the highest purity available, and used without further purification. Undoped BiVO4 photoanodes were fabricated by modification of a MOD method previously reported.28 Briefly, bismuth nitrate pentahydrate (0.2 mol L−1) was dissolved in acetic acid (1.5 mL, BDH), and vanadyl acetylacetonate (0.2 mol L−1) was dissolved in acetylacetone (10 mL, Merck). These solutions were stirred at room temperature for 30 minutes. FTO (fluorine-doped tin oxide) glass (2.5 × 2.5 cm, TEC 15, Hartford Glass Co.) was cleaned by sonication in de-ionised water (ELGA PURELAB Option-Q, 18.2 MΩ cm at 25 °C) then acetone (VWR). After sonication, the cleaned glass was heated at 500 °C for 30 minutes to remove all organic residues.BiVO4 films were prepared by depositing the precursors by spin-coating. Spin-coating parameters were set at 1000 rpm for 20 s for each layer of deposition. For each layer, a small piece of the substrate was covered with a piece of tape to leave blank FTO for electrical connection. After each layer was deposited by spin-coating, the film was calcined at 450 °C for 30 min. This spin-coat/calcine procedure was repeated 15 times. After depositing the 15th layer, the coated glass was calcined at 450 °C for 10 hours to ensure formation of the monoclinic-scheelite structure.
Characterisation
X-ray diffraction (XRD) was conducted using a micro-focus Bruker GADDS powder X-ray diffractometer with a monochromated Cu Kα (1.5406 Å) source. UV-vis spectra were obtained using a Perkin Elmer UV-vis spectrometer (Lambda 25). SEM imaging was carried out with a LEO 1525 scanning emission microscope (field emission gun SEM with acceleration voltage of 5 kV).Photoelectrochemical (PEC) measurements were carried out in a home-made PTFE cell with quartz windows on front and back sides. A three-electrode configuration was used, including a platinum mesh counter electrode, a reference electrode (Ag/AgCl/saturated KCl, 0.197 VNHE at 298 K; Metrohm) and the BiVO4 working electrode. Unless otherwise stated, all PEC and transient absorption spectroscopic measurements were carried out via back-side (substrate–electrode) illumination. The scan rate was 10 mV s−1 in the current–potential (I–V) measurement. Potassium phosphate (KPi) buffer (0.1 mol L−1 K2HPO4 and KH2PO4) was employed to maintain a constant pH of 6.7. Na2SO3 (1 mol L−1) and AgNO3 (6 mmol L−1) were used as the hole scavenger and electron scavenger, respectively. The applied potential used in this paper was converted to be versus the reversible hydrogen electrode (RHE) using the Nernst equation (eqn (1)),
| VRHE (V) = VAg/AgCl (V) + 0.0591 × pH + V0Ag/AgCl | (1) |
The light source used for photocurrent and IPCE measurements was a 75 W ozone-free xenon lamp (Hamamatsu) coupled with a monochromator (white light intensity: ∼100 mW cm−2). An Autolab potentiostat (PGSTAT 12 equipped with FRA2 module) was used to record all PEC and impedance data with Nova software. In incident-photon-to-current efficiency (IPCE) measurements, the intensity of the monochromated light was measured using an optical power meter (PM 100, Thorlabs) with a power sensor (S120UV, Thorlabs). The IPCE values were calculated using the following equation (eqn (2)),29
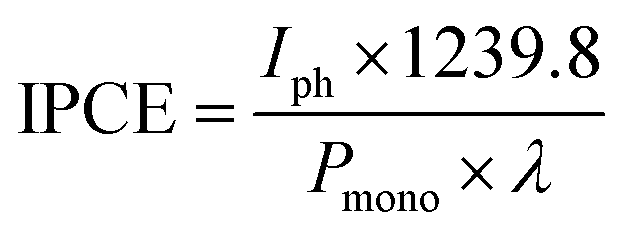 | (2) |
Transient photocurrent (TPC) measurements were carried out by fixing constant applied bias to the sample and then measuring the current response under chopped light conditions. The light on/off condition was controlled by a home-programmed shutter with a time interval of 10 s.
Electrochemical impedance spectroscopy (EIS) measurements in the dark were conducted to determine the flat-band potential (VFB) of BiVO4. The frequencies were between 0.01 Hz and 100 kHz. The Randles equivalent circuit (shown in Fig. S4†) was used to analyze the impedance data using Zview software (Scribner Associates). The VFB was calculated using the Mott–Schottky equation (eqn (3)),
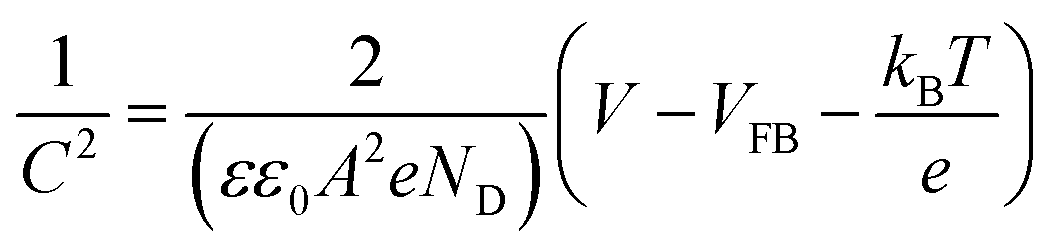 | (3) |
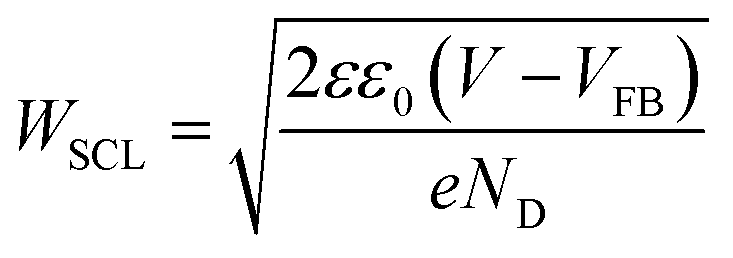 | (4) |
Transient absorption spectroscopy (TAS)
The transient absorption spectrometer setup has been described previously.32 Briefly, the transient absorption spectrometer consists of a Nd:YAG laser (Big Sky Laser Technologies, Ultra CFR Nd:YAG laser system, 6 ns pulse width) at 355 nm (3rd harmonic) to excite the band gap of BiVO4 photoanodes; the frequency of the laser flash was 0.33 Hz; the laser intensity was adjusted to be 100 μJ cm−2 using neutral density filters (Comar Instruments), except during the excitation intensity study, during which the laser intensity ranged from 2 μJ cm−2 to 800 μJ cm−2. The probe light source was a 100 W Bentham IL1 tungsten lamp equipped with a monochromator (OBB-2001, Photon Technology International). The transmitted probe light was filtered by a number of long-pass filters and a band-pass filter (Comar Instruments) in order to filter the scattered laser light. A silicon photodiode (Hamamatsu S3071) was used to detect transmitted photons. Collected data were processed in an amplifier (Costronics), and then were recorded with an oscilloscope (Tektronics TDS 2012c) on the timescale of μs–ms, and with a DAQ card (National Instruments, NI USB-6211) on the timescale of ms–s. Each transient absorption decay was recorded by averaging over 300 laser pulses. All data were acquired by home-programmed software on the Labview platform.Results
Structural characterization (SEM and XRD) indicated that the BiVO4 films employed in this study comprise dense, rather flat films (particle size ∼100 nm, thickness 450–500 nm) with a monoclinic-scheelite crystal phase (see ESI Fig. S1 and S2†), consistent with a previous study of analogous BiVO4 films.28 According to the thickness determined from SEM images and the absorbance measured from UV-vis spectroscopy, the light penetration depth at 355 nm excitation was calculated to be ∼270 nm in the BiVO4 photoanode (see ESI Section 3 and Fig. S3†).Fig. 1 shows the current–voltage performance of a typical BiVO4 photoanode employed in this study, consistent with a previous study reported by Sayama et al.28 The dark current onset for water oxidation is 1.9 VRHE, which is indicative of a large overpotential for dark water oxidation on BiVO4. At 1.2 VRHE, the photocurrent is 0.86 mA cm−2. The corresponding incident photon-to-current efficiency spectrum (IPCE, shown in ESI Fig. S3†) shows an onset at 500 nm, corresponding to the BiVO4 absorption edge. The photocurrent onset is ∼0.8 VRHE.
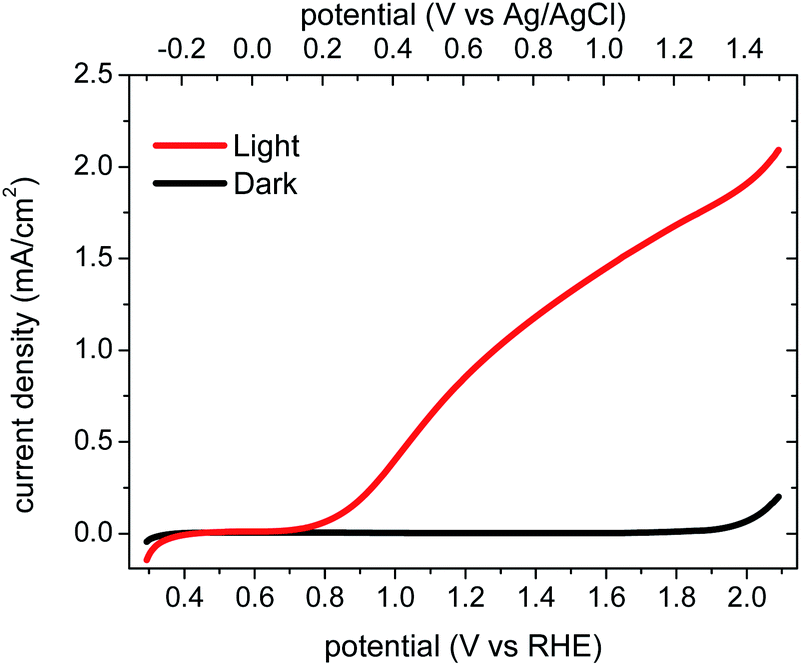 | ||
| Fig. 1 The current density of a BiVO4 photoanode, measured in 0.1 M KPi buffer (pH 6.7), is shown as a function of the applied bias versus RHE and Ag/AgCl reference potentials. Current density measured under illumination (red line) is compared to dark conditions (black line). Light intensity: approximately 100 mW cm−2. | ||
Mott–Schottky analysis using electrochemical impedance spectroscopy was employed to monitor space charge layer formation in these photoanodes (see ESI, Fig. S4†). The flat-band potential (VFB) was determined to be ∼0.1 VRHE, which is 700 mV more negative than the onset of photocurrent generation. This analysis also yielded a donor density of 1018 cm−3, similar to that reported previously for ‘undoped’ BiVO4 films (i.e. without additional intentional dopants).16 This donor density value was used to calculate the width of the space charge layer (WSCL) as a function of electrical bias using eqn (4), as further discussed below (see Fig. 5). The relatively low donor density measured for these undoped BiVO4 photoanodes results in a rather wide space charge layer (e.g. at 1.2 VRHE, WSCL was calculated to be ∼90 nm),33 significantly wider than typically reported in, for example Si-doped hematite photoanodes.34
Using transient absorption spectroscopy, charge carriers photogenerated in the BiVO4 photoanode can be directly monitored by measuring the change in the intensity of a transmitted monochromatic probe light as a function of time after pulsed band-gap excitation. In order to understand the photogenerated holes' behavior during the process of water oxidation on BiVO4, it is first necessary to identify the transient absorption signal of these holes. Employing electron/hole scavengers or applying anodic/cathodic bias can enable the identification of charge carriers' transient absorption signals by modulating their lifetimes and concentrations. Previous transient absorption studies have shown that electron/hole scavengers change the kinetics of charge carriers significantly in WO3 and TiO2 films;26,35 however, kinetics of photogenerated holes in α-Fe2O3 were not affected by methanol (a hole scavenger) without applied bias.20
Fig. 2a compares the transient absorption data collected for a BiVO4 photoanode in de-ionised water, AgNO3 solution, and Na2SO3 solution with no applied electrical bias, measured at 550 nm. We note that these films exhibited significant optical light scattering; the resultant negative scatter signal precludes the collection of transient absorption data for <10 μs time delays in our apparatus. The transient absorption spectra measured under the same conditions are shown in Fig. 2b at a time delay of 50 μs. The observed enhancement of the transient absorption signal amplitude and the increased lifetime of charge carriers in the presence of the electron scavenger (AgNO3) suggest that photogenerated holes are probed in this spectral region (500–900 nm). This assignment is supported by a significant decrease in the signal amplitude and lifetime on the addition of the hole scavenger Na2SO3. The assignment of transient signals in this spectral region, peaking at 550 nm, to photogenerated holes is also supported by the transient absorption study of BiVO4 with applied electrical bias (see below), and with a previous transient absorption study of BiVO4 employing FeCl3 as an electron scavenger.22 Absorption maxima for photoinduced holes in this spectral region have also been observed for other metal oxide photoelectrodes.20,25,26,35 No signals were observed that could be clearly assigned to electrons between 500 and 900 nm, suggesting that electrons may absorb in the near-IR region, as is observed in some other metal oxide photoelectrodes.26,35 In any case, the dynamics of photogenerated holes and their reaction with water are the primary focus of this study; our assignment indicates these holes can be monitored most readily at 550 nm.
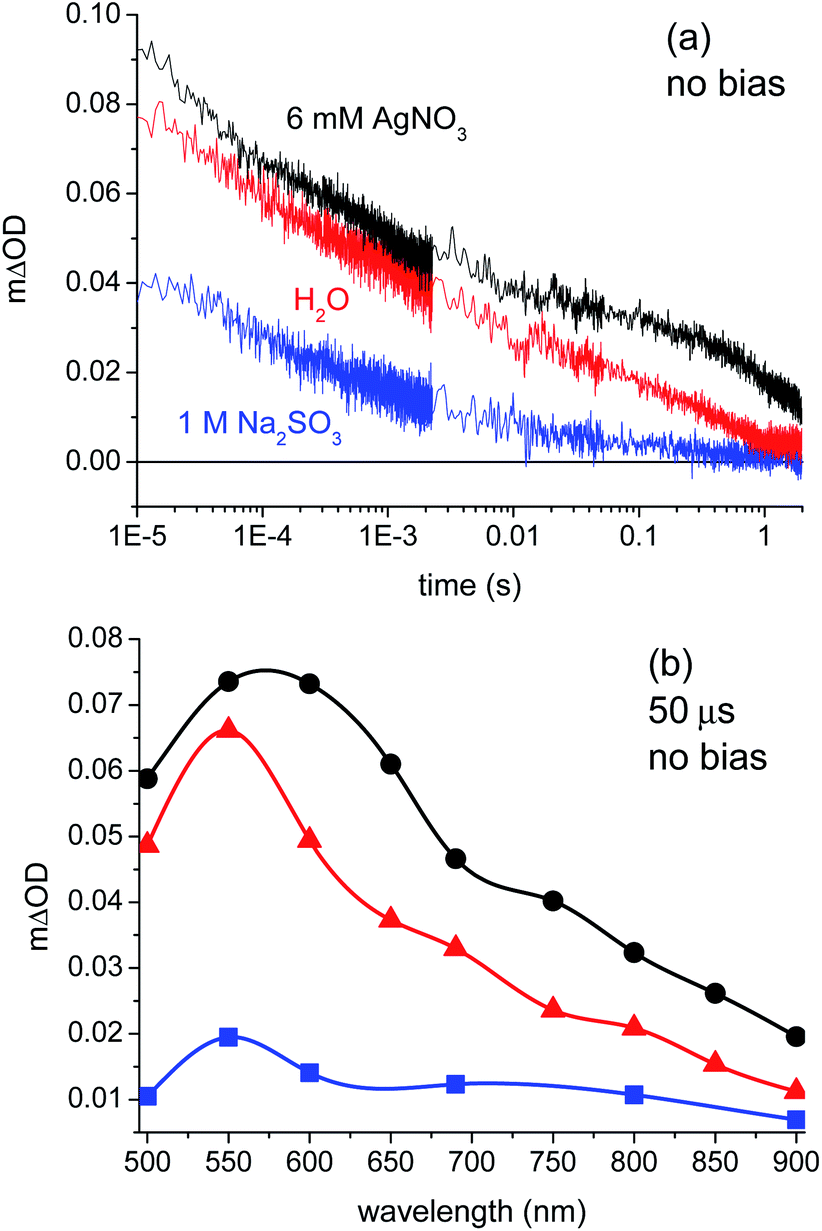 | ||
| Fig. 2 (a) Transient absorption decays of a BiVO4 photoanode measured in 6 mM AgNO3 (black, electron scavenger), de-ionised water (red) and 1 M Na2SO3 (blue, hole scavenger), probing at 550 nm. (b) Transient absorption spectra in AgNO3, de-ionised water and Na2SO3 (color matches (a)), recorded 50 μs after laser excitation (355 nm, 100 μJ cm−2, 0.33 Hz). | ||
We turn now to data collected under applied electrical bias. The decay dynamics of photogenerated holes, probed at 550 nm, in a BiVO4 photoanode as a function of applied bias from 0.1 VRHE (flat-band potential, VFB) to 1.5 VRHE are shown in Fig. 3a. At VFB, the small transient absorption signal decays to zero within 20 ms, assigned to charge recombination occurring primarily on timescales faster than our instrument response, consistent with a previous study of α-Fe2O3 photoanodes.20 The amplitude and decay time constant of the photoinduced hole signal both increase with increasing positive (anodic) applied bias throughout the wavelength range studied (see ESI Fig. S5†). This is consistent with our assignment of these transient absorption signals to photogenerated holes, with applied anodic bias increasing the yield of these holes due to reduction of electron/hole recombination losses. We note that the transient kinetics of the unbiased film in the absence of scavengers is closest to that observed under modest anodic bias (∼0.6 VRHE), indicative of the presence of significant band bending in these BiVO4 electrodes in water in the absence of any applied bias or scavengers. The dark open circuit potential of the unbiased BiVO4 photoanode was measured to be 0.6 VRHE, which is in agreement with these results.
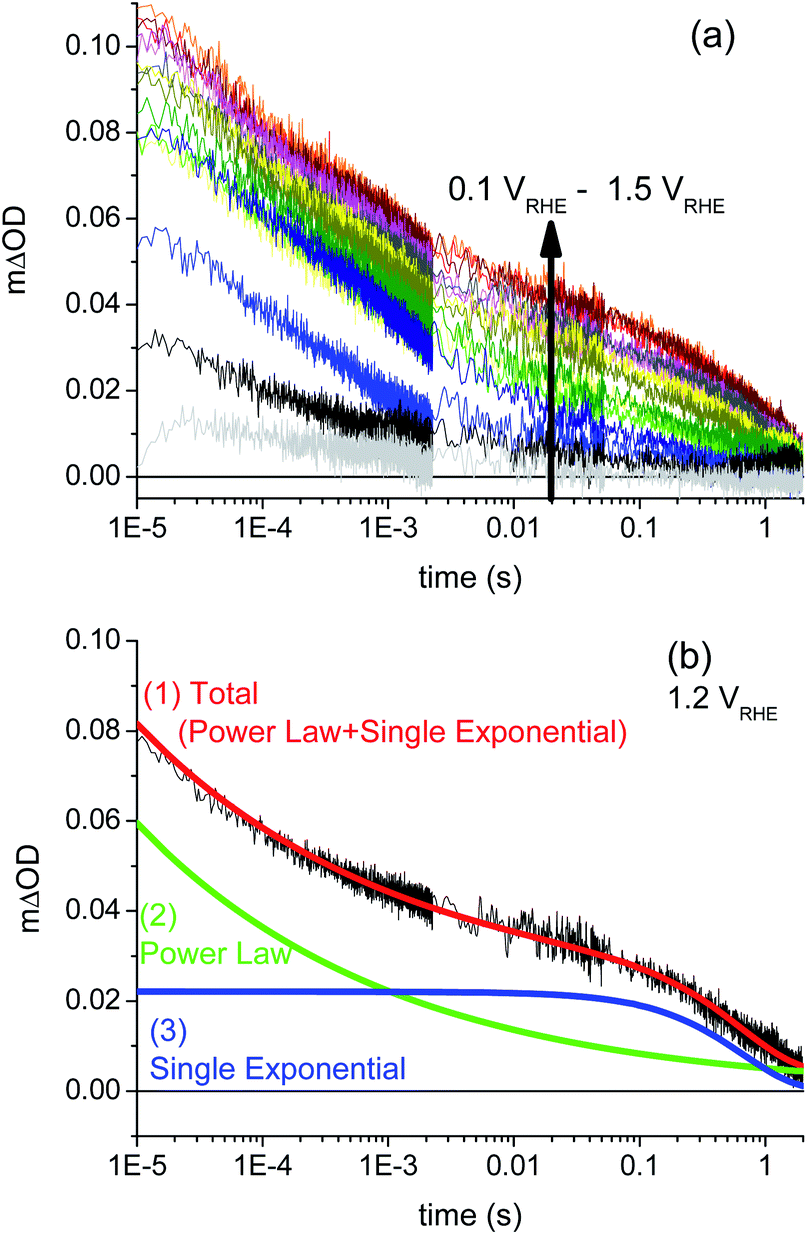 | ||
| Fig. 3 (a) Dynamics of photogenerated holes (probed at 550 nm) in the BiVO4 photoanode as a function of applied bias, measured from 0.1 VRHE (corresponding to flat-band conditions) to 1.5 VRHE, at intervals of 0.1 V. Data recorded in a three-electrode PEC cell with KPi electrolyte; other conditions as for Fig. 2. (b) The fitting (red) to a decay measured under 1.2 VRHE applied bias, with a combination of a power law (green) and single exponential (blue) functions. | ||
At strong positive (anodic) bias, a biphasic decay is observed, with one phase on microsecond to millisecond timescales (defined herein as the fast phase), and a slower decay phase on millisecond to second timescales. This biphasic behavior under anodic bias is similar to that which we have reported previously for TiO2 and α-Fe2O3 photoanodes,20,21 where the faster μs–ms decay phase was assigned to bimolecular electron/hole recombination following photoexcitation, and the slower ms–s phase to holes localised in the space charge layer. The similarity with the data reported herein suggests similar assignments of these two phases can be made for the BiVO4 photoanodes studied herein.
Support for assignment of the faster phase to electron/hole recombination comes from the laser intensity dependence of the transient absorption decays at 1.2 VRHE, as shown in Fig. 4a. It is apparent that the decay kinetics of the slow decay phase (ms–s) are almost independent of the excitation intensity (see ESI, Fig. S8†), whereas the fast phase becomes more pronounced and shorter lived as the laser intensity is increased (see also ESI Fig. S7†). The increased dominance and acceleration of the fast phase with increased laser intensity is analogous to behavior we have observed for α-Fe2O336 and TiO235 photoanodes, and is consistent with the assignment of this phase to the bimolecular recombination of photogenerated electrons and holes. The saturation in the amplitude of the slow transient absorption decay phase under high excitation intensities (>400 μJ cm−2) is attributed to the typical non-linear dependence of bimolecular recombination on charge carrier density (with an increasing proportion of this recombination occurring on sub-microsecond timescales not reported herein). We note that the measured amplitude of the slow phase (measured on millisecond timescales) saturates at 5 × 10−5 ΔOD (absorption) units as the laser intensity is increased, emphasising the high detection sensitivity required to monitor this slow decay phase.
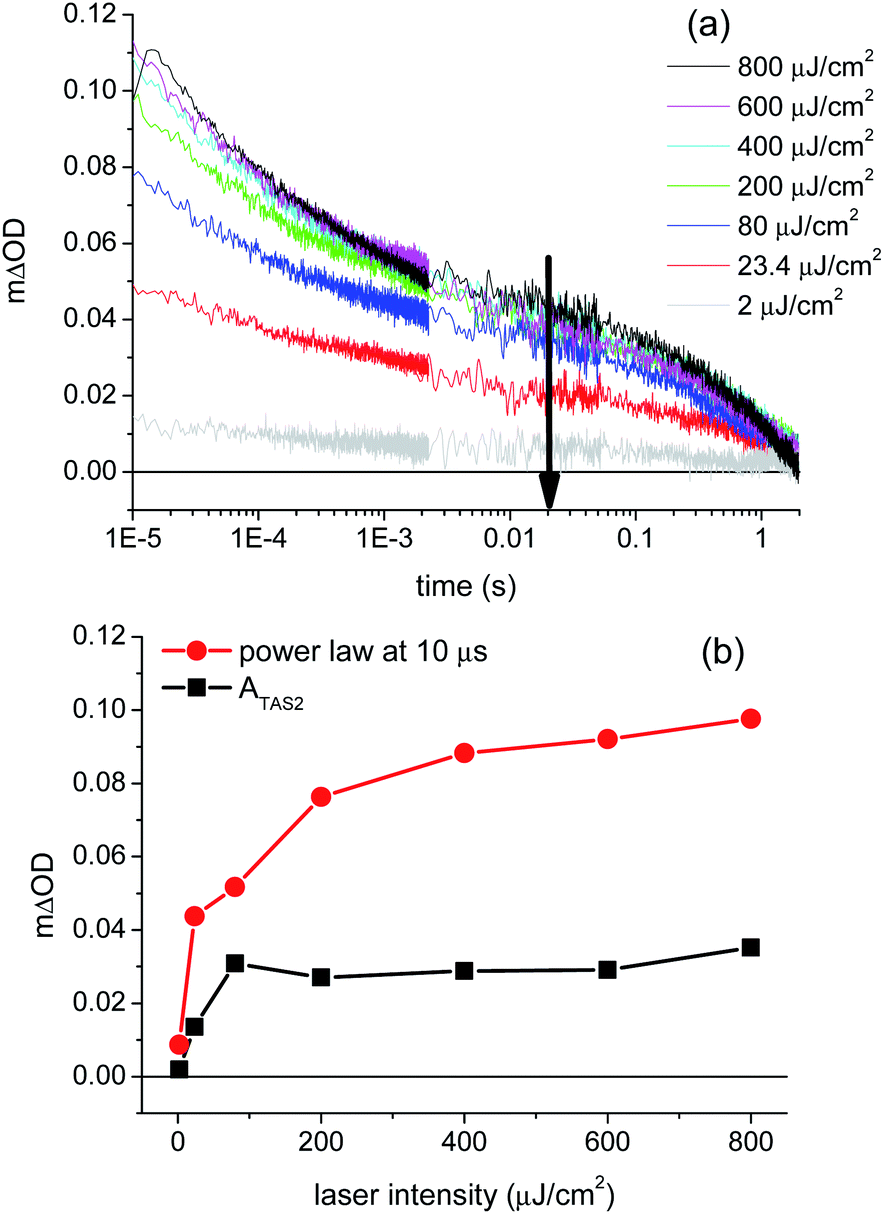 | ||
| Fig. 4 (a) Transient absorption decays of a BiVO4 photoanode as a function of laser excitation intensity, ranging from 2 μJ cm−2 (grey) to 800 μJ cm−2 (black), at 1.2 VRHE. The arrow in the figure indicates decreasing laser intensity. (b) Amplitude from the fit of the μs–ms phase (power law; red dots) and the amplitude of the ms–s phase (ATAS2 of the exponential function; black squares) as a function of laser excitation intensity. | ||
The transient absorption decays such as those shown in Fig. 3a and b are well fitted by a combination of a power law decay (consistent with bimolecular electron/hole recombination, see ESI Fig. S6†) and a slower, single exponential function, as detailed in eqn (5):
| ΔOD(t) = at−b + ATAS2e−t/τ(TAS2) | (5) |
It is apparent from Fig. 3 and 5 that the amplitude ATAS2 of the slow, exponential TAS decay phase increases with increasing anodic bias beyond a threshold voltage of ∼0.2 VRHE. The lifetime τ(TAS2) of this exponential decay phase also increases with anodic potential, as is apparent from Fig. 3, and is discussed further below (see Fig. 7). In Fig. 5, we overlay the amplitude of this decay phase (ATAS2, filled squares) as a function of applied bias with the photocurrent density measured under white light irradiation (dashed line), and with the width of the space charge layer determined by our electrochemical impedance analysis (solid line). Following our previous studies of hematite photoanodes, this amplitude is assigned as an assay of the yield of photogenerated holes which avoid initial recombination with BiVO4 electrons and are thus able to accumulate at the photoanode surface. It is apparent that the yield of these long-lived holes correlates with the width of the space charge layer; both have an onset potential of 0.2 VRHE. It is also striking that for modest applied potentials, 0.2–0.7 VRHE, these surface-localised holes do not result in significant photocurrent generation (i.e. water oxidation). This implies the presence, at these potentials, of a process that competes with water oxidation, which we consider below.
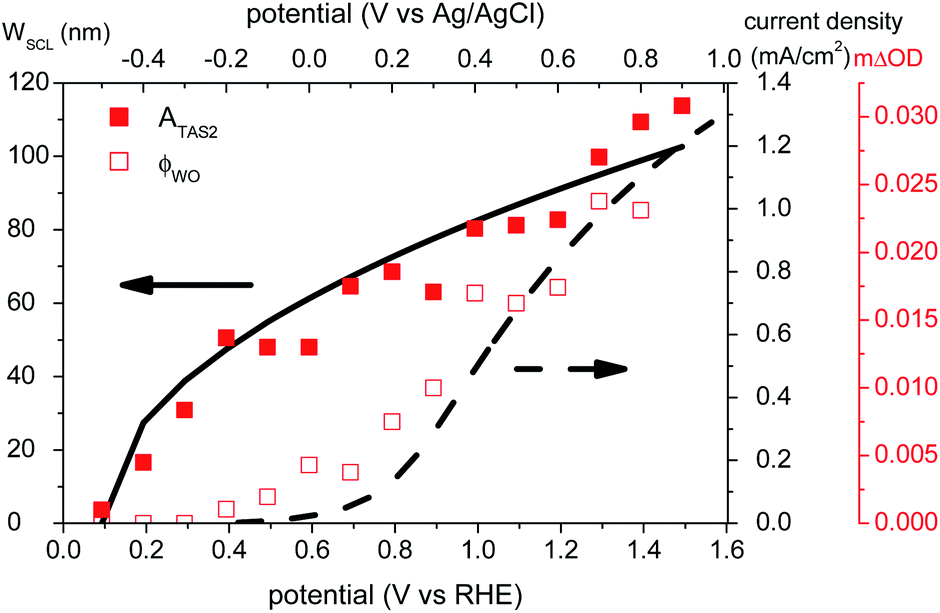 | ||
| Fig. 5 Correlation of transient absorption amplitude of long-lived photogenerated holes (ATAS2, red solid squares, obtained from fitting with an exponential decay) with the width of the space charge layer (black solid line), and correlation of yield of water oxidation determined from the transient absorption data by eqn (7) (ϕWO, red empty squares) with photocurrent density (black dashed line). | ||
We turn now to complementary transient photocurrent measurements18 and their correlation with the transient absorption data reported above. Fig. 6a presents the result of a typical chopped light transient photocurrent measurement under modest anodic bias (0.8 VRHE). When the light is switched on, a positive transient photocurrent is observed; when it is switched off, an analogous negative transient is observed, in qualitative agreement with a previous BiVO4 report.13 These positive and negative photocurrent transients have been associated with accumulation of the holes in the BiVO4 space charge layer under prolonged irradiation and the recombination of bulk electrons with these holes,13 in agreement with analogous studies of α-Fe2O3 photoanodes.18,19,23 In particular, the negative transient observed following light switch-off has been assigned to recombination of bulk BiVO4 electrons with holes accumulated in the space charge layer during irradiation, resulting in a back flow of electrons into the photoanodes.13
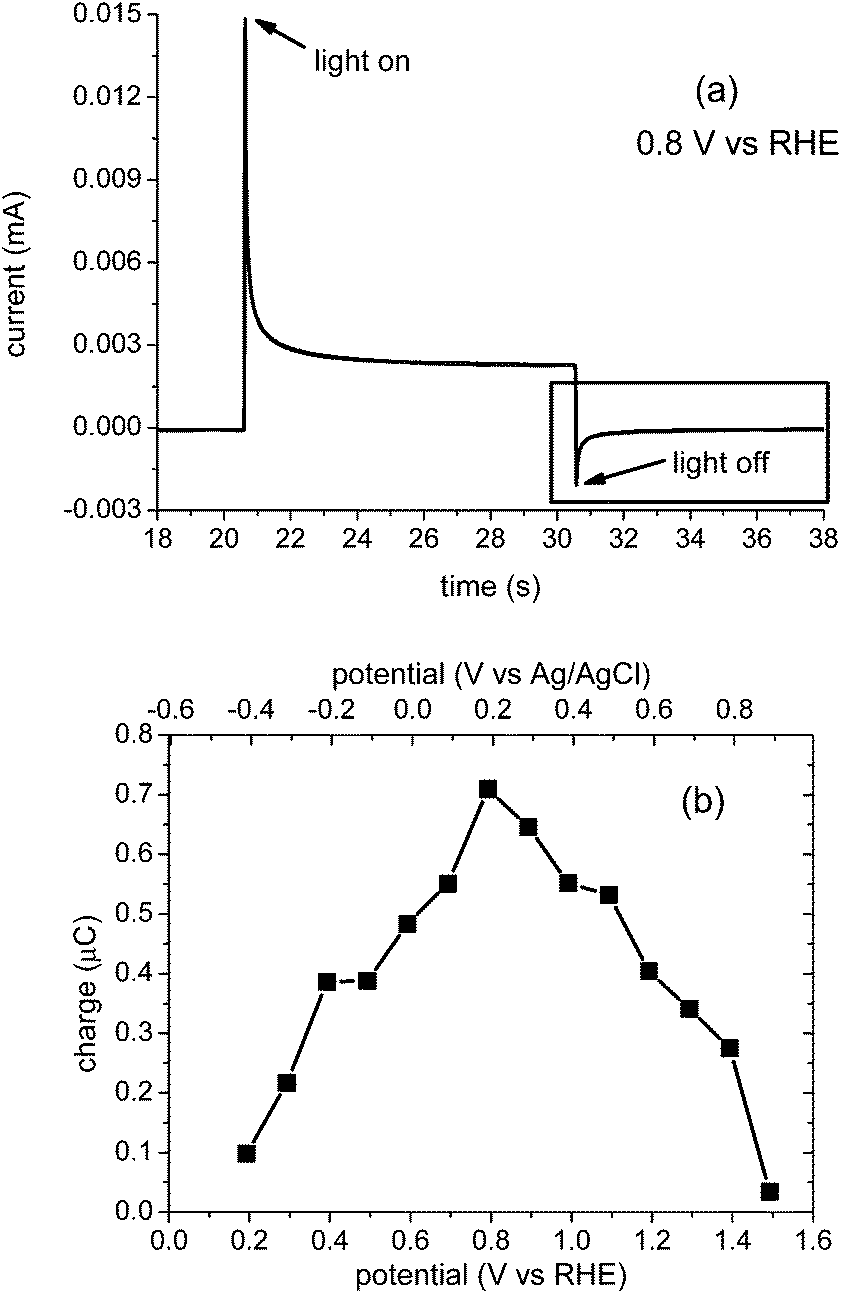 | ||
| Fig. 6 (a) Chopped light transient photocurrent measured at 0.8 VRHE. (b) Charge due to surface accumulated holes which undergo ms–s recombination, as a function of applied bias, obtained by integration of the negative transient current (black box in (a)). | ||
Analogous chopped light photocurrent data were collected for applied bias between 0.2 and 1.5 VRHE. The negative transients fitted reasonably to single exponential decays, shown in eqn (6),19
| I(t) = I0 + ATPCe−t/τ(TPC) | (6) |
Integration of the negative transient current quantifies the negative charge associated with this back electron/hole recombination process, as shown in Fig. 6b. It is apparent that losses associated with this back electron/hole recombination are largest for potentials around the photocurrent onset (0.8 VRHE), and negligible for potentials ≤0.2 VRHE and ≥1.5 VRHE. It is also apparent that the potential range where this recombination phase results in the largest losses corresponds to the range where we observe high yields of long-lived holes but low photocurrent densities (see ESI Fig. S10†).
The time constants of the negative photocurrent transients decays (τ(TPC), red circles) are shown as a function of applied bias in Fig. 7, increasing from 10 ms to ∼1 s with increasing anodic bias. The time constants of these ‘back electron/hole recombination’ transients appear to be very similar to the decay times of the slow decay phase observed in the transient absorption data, τ(TAS2), black squares in Fig. 7, as we discuss below.
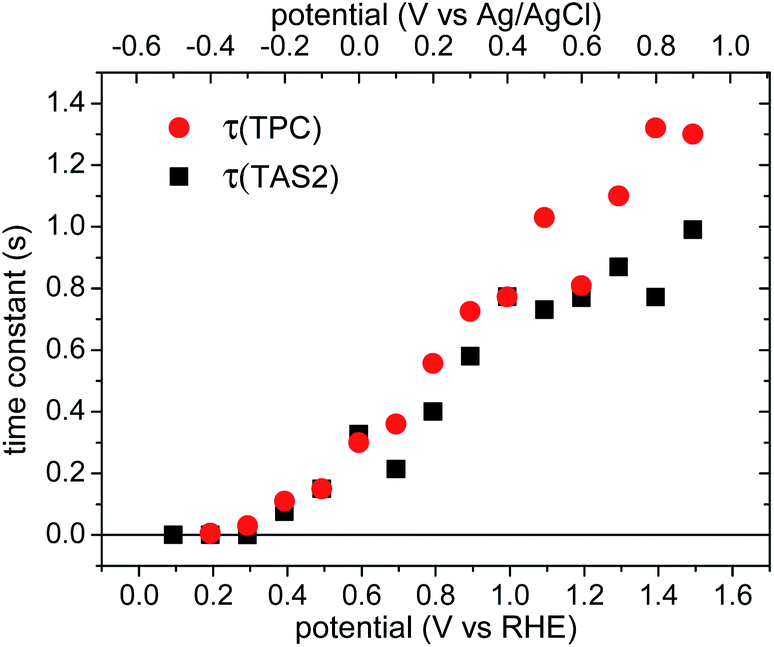 | ||
| Fig. 7 Comparison of the time constants as a function of applied bias obtained from transient absorption spectroscopy and transient photocurrent. Red dots: τ(TPC), the time constants of negative transient current in the chopped light TPC measurements; black squares: τ(TAS2), slow phase time constants obtained from fitting the transient absorption slow decay phase with an exponential function. | ||
Discussion
Recombination dynamics on μs–ms timescales
Using chemical scavengers and applied bias, the transient absorption signal peaking at 550 nm has been assigned to photoinduced BiVO4 holes, consistent with a previous study.22 We first of all consider the decay dynamics of this transient absorption on the μs–ms timescale. The slower, ms–s decay phase observed under strong anodic electrical bias is discussed in the subsequent section.Our initial assignment of the fast, μs–ms power law decay phase to bimolecular electron/hole recombination, based on analogy with comparable data obtained for α-Fe2O3 and TiO2 films,20,21 is strongly supported by the excitation intensity study shown in Fig. 4a. In this study, the initial (10 μs) amplitude of the fast phase the transient absorption signal significantly increases with increasing laser intensities between 0 and ∼100 μJ cm−2 but saturates above 100 μJ cm−2. An excitation intensity of >100 μJ cm−2 corresponds to a density of absorbed photons of 1018 cm−3, similar to the donor density of these films (∼1018 cm−3, calculated from Mott–Schottky analysis). The decay kinetics are slightly accelerated by high excitation intensities, due to elevated concentrations of photogenerated holes (and electrons) resulting in faster bimolecular recombination. It should be noted that the acceleration of decay kinetics with increasing excitation intensity is not as significant as has been reported for α-Fe2O3 and TiO2 photoanodes.21,36
Yield of long-lived BiVO4 holes
We now consider the slower, exponential transient absorption decay phase (ms–s) observed under anodic bias. Fig. 5 shows the transient absorption amplitude of long-lived holes after laser excitation as a function of applied bias (red solid squares), overlaid on the width of the space charge layer (WSCL, black solid line) determined from our Mott–Schottky impedance analysis. The relatively large width of this space charge layer (e.g. ∼90 nm at 1.2 VRHE) is in agreement with a previous impedance analysis of undoped BiVO4,37 and consistent with the relatively low doping density compared to intentionally doped metal oxide photoanodes.34,36 It is apparent that there is a quantitative correlation between this assay of long-lived photogenerated holes and the space charge layer width. This strongly suggests that the electric fields present in the space charge layer are essential for driving the spatial separation of initially generated electrons and holes, and thereby the generation of long-lived holes which accumulate at the electrode surface.It is also apparent from Fig. 5 that this transient absorption assay of long-lived holes does not correlate with the photocurrent density (black dashed line). Significant long-lived hole signals are observed in BiVO4 at potentials cathodic of the photocurrent onset potential Von (0.8 VRHE). Since photocurrent is indicative of water oxidation, as has been shown previously for BiVO4 photoanodes,15 the observation of long-lived holes at V < Von suggests that there must be a competing pathway that reduces the yield of water oxidation, particularly at modest applied bias. In the following section, we address the identity of this competing pathway.
Kinetic competition between water oxidation and back electron/hole recombination
We have observed that the decay kinetics of the slow transient absorption decay phase correlates with the recovery time of the negative transient current assigned to back electron/hole recombination, as illustrated in Fig. 6a. The transient absorption signal on ms–s timescales monitors the density of long-lived holes. The observation that the negative current transient decays with the same time constant as the decay of this hole density supports our assignment of this current transient to back electron transfer into the photoanode. This back electron transfer is driven by recombination of bulk BiVO4 electrons with holes accumulated in the space charge layer, analogous to recent analyses of hematite photoanodes.18,19,23We now apply a kinetic model to the transient absorption results to quantify the proportion of long-lived photogenerated holes which give rise to water oxidation versus those which decay due to back electron/hole recombination, as shown in Scheme 1. The water oxidation hole yield, ϕWO(V), assuming a 100% Faradaic efficiency consistent with previous reports,12,16 can be calculated from,
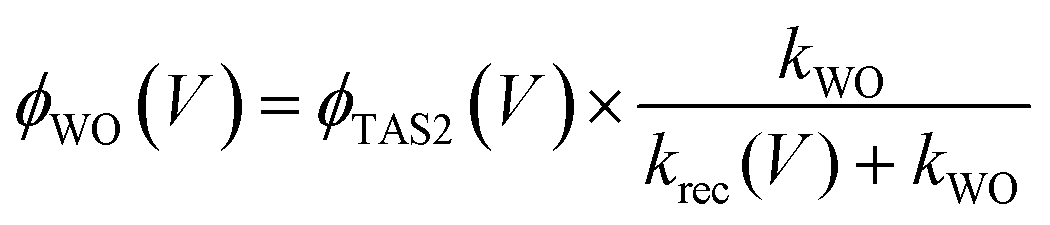 | (7) |
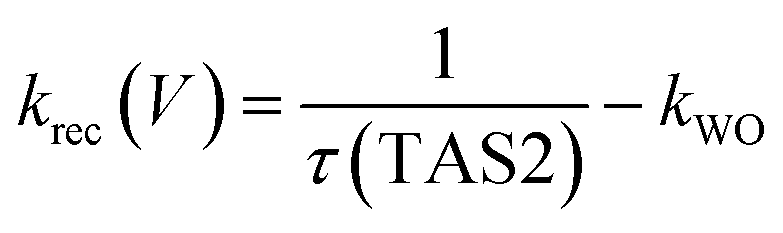 | (8) |
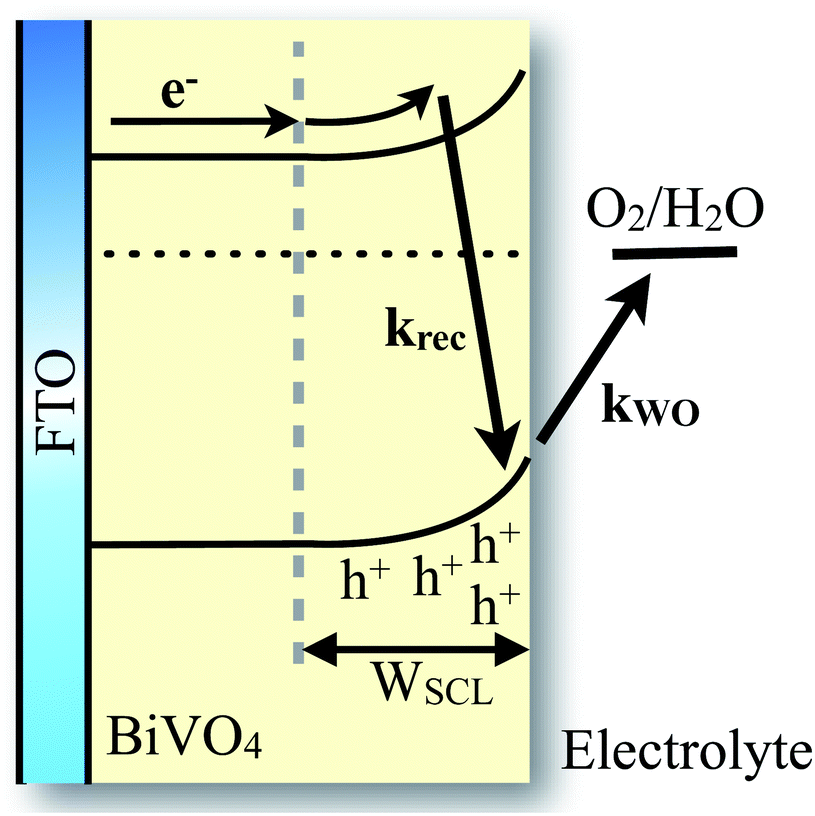 | ||
| Scheme 1 Schematic representation of the photo-induced processes considered in the kinetic model of photogenerated charges in an undoped BiVO4 photoanode. The back electron/hole recombination is in kinetic competition with water oxidation, on ms–s timescales. | ||
The transient absorption decay kinetics of long-lived photogenerated holes saturates (time constant ∼0.8 s) at applied bias more positive than 1.3 VRHE, as shown in Fig. 7. Over this potential range, losses from back electron/hole recombination tend to zero (Fig. 6b). This suggests that over this potential range, water oxidation is dominant, such that kWO ∼ 1.3 s−1.
The yield of water oxidation, ϕWO, determined from analysing the slow transient absorption decay phase by eqn (7) and (8) is shown as a function of applied bias in Fig. 5 (empty squares). It is apparent that the yield of water oxidation determined from this simple kinetic model correlates remarkably well with the photocurrent density. In particular, this kinetic analysis of our transient absorption data suggests that almost all long-lived holes recombine without contributing to water oxidation between 0.1 VRHE (VFB) and 0.8 VRHE (Von). Back electron/hole recombination decreases with increasing anodic bias after Von and is strongly suppressed at strong anodic potentials. This observation is consistent with our transient photocurrent data, which indicate that losses resulting from back electron/hole recombination are maximal around the photocurrent onset (0.8 VRHE), and become smaller for higher applied potentials. For potentials >1.2 VRHE, the slow decay phase of photogenerated holes is dominated by water oxidation.
We have previously reported that the yield of long-lived holes observed with α-Fe2O3 and TiO2 photoanodes shows strong correlation with photocurrent density,36,38 although these studies did not consider the bias dependence of the lifetime of these long-lived holes, nor the impact of back electron/hole recombination as discussed herein. We have recently reported an analogous study of Si-doped, nanostructured α-Fe2O3 photoanodes23 where we include consideration of such back electron/hole recombination, and demonstrate that the model reported herein for BiVO4 is also valid for other photoanode materials such as α-Fe2O3, consistent with previous PEC analyses of α-Fe2O3 electrodes.18,19 We note however that the potentials relative to flat band required to generate long-lived holes, and to suppress back electron/hole recombination, differ between the results for BiVO4 reported herein and those we have reported for Si-doped α-Fe2O3. For example, for the BiVO4 photoanodes long-lived holes are observed at potentials immediately anodic of flat band, whereas for Si-doped α-Fe2O3 such long-lived holes are only observed at potentials 300 mV more positive than flat band, potentially due to the much narrower space charge layer in the doped α-Fe2O3 compared to the BiVO4 photoanodes studied herein. Such comparisons will be addressed in more detail elsewhere.
The kinetics of the slow TAS decay phase at strong anodic bias, assigned to water oxidation, are observed to be independent of excitation intensity, and therefore hole density (Fig. 4b and ESI Fig. S8†). This observation suggests that under the experimental conditions employed, this water oxidation process does not involve a concerted, multi-hole water oxidation, but rather the sequential single-hole water oxidation. As we have discussed previously, the single-hole oxidation of water by metal oxide holes such as BiVO4, α-Fe2O3 and TiO2 may be enabled by the highly oxidizing nature of such valence band holes.39 It should however be noted that continuous solar irradiation may result in substantially higher densities of accumulated holes than those achieved by the pulsed laser excitation employed herein, with therefore potentially greater opportunities for concerted, multi-hole reactions.
We now consider the mechanism of the back electron/hole recombination which we observe to be in kinetic competition with water oxidation. It should be noted that electrons cannot reach the semiconductor–liquid interface without being thermally activated over or tunneling across the space charge layer (or vice versa for holes moving into the electrode bulk). Tunneling across the SCL is unlikely due to the relatively wide SCL in the photoanodes studied herein (∼80 nm at 0.8 VRHE). More likely, the relatively slow timescales of back electron/hole recombination (100 ms) compared to bulk electron recombination (typically reported on picosecond timescales for metal oxides) can be attributed to the requirement for thermal activation across the SCL. For example, a 300 mV barrier height is equivalent to 12 kBT at room temperature, resulting in a retardation of recombination dynamics by e−12 or 1/200000 fold. It thus appears more likely that this back-electron recombination proceeds via thermal activation.
The formation of a space charge layer at the semiconductor–liquid interface is widely recognized as being critical in reducing recombination losses in photoelectrodes. The results we present herein suggest that two distinct, space-charge dependent, recombination processes must be considered for the BiVO4 photoanodes studied herein. The space charge layer facilitates initial charge separation, leading to generation of long-lived holes with a yield that scales with the width of the space charge layer. However, for potentials only modestly anodic of flat band, the space charge layer field is insufficient to block the slow back recombination of bulk electrons with these long-lived holes localized in the space charge layer. This back electron/hole recombination can be avoided by significantly deep band bending across the space charge layer with strong anodic bias. In the BiVO4 photoanodes employed in our studies, the potential required to prevent back electron/hole recombination appears to be ∼600 mV greater than that required to achieve separation of the initially generated charge carriers, and is therefore the key reason for the high anodic voltage required for photocurrent generation. As such, the results reported herein suggest that blocking the back electron/hole recombination should be prioritised in order to improve the water oxidation efficiency of BiVO4 photoanodes. This could be achieved by increasing barriers to this recombination (by increasing the space charge layer band bending or introducing an additional junction) or using surface catalysts to increase the water oxidation rate constant.
Conclusion
In this paper we have focused on the study of water oxidation and recombination dynamics of photogenerated holes in undoped BiVO4 photoanodes using transient absorption spectroscopy and photoelectrochemical methods. The transient absorption signals of the holes on microsecond to second timescales were assigned using an electron scavenger, a hole scavenger, and applied electrical bias. A broad absorption of holes in BiVO4 photoanodes was observed from 500 nm to 900 nm, peaking at 550 nm. However, charge recombination on ms–s timescales occurs due to back electron/hole recombination. This slow recombination kinetically competes with water oxidation, thus limiting the photoanode efficiency. These results suggest that future material design and optimisation should consider avoiding back electron/hole recombination in BiVO4 photoanodes in order to enable efficient water oxidation at less anodic applied potentials.Acknowledgements
We thank the European Science Foundation (project Intersolar 291482) for funding. F.L.F. thanks the Swiss National science Foundation (project: 140709) and A.R. thanks the European Commission Marie Curie CIG (303650). The authors also thank Dr Andreas Kafizas for help with XRD measurements and Dr Piers Barnes for helpful discussions.Notes and references
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- Y. Park, K. J. McDonald and K.-S. Choi, Chem. Soc. Rev., 2013, 42, 2321–2337 RSC.
- H. Ye, H. S. Park and A. J. Bard, J. Phys. Chem. C, 2011, 115, 12464–12470 CAS.
- H. S. Park, K. E. Kweon, H. Ye, E. Paek, G. S. Hwang and A. J. Bard, J. Phys. Chem. C, 2011, 115, 17870–17879 CAS.
- A. J. E. Rettie, H. C. Lee, L. G. Marshall, J.-F. Lin, C. Capan, J. Lindemuth, J. S. McCloy, J. Zhou, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2013, 135, 11389–11396 CrossRef CAS PubMed.
- F. F. Abdi, N. Firet and R. van de Krol, ChemCatChem, 2013, 5, 490–496 CrossRef CAS.
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781–1787 CAS.
- P. Chatchai, Y. Murakami, S. Y. Kishioka, A. Y. Nosaka and Y. Nosaka, Electrochem. Solid-State Lett., 2008, 11, H160–H163 CrossRef CAS PubMed.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef CAS.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- J. Yang, D. Wang, X. Zhou and C. Li, Chem.–Eur. J., 2013, 19, 1320–1326 CrossRef PubMed.
- J. A. Seabold and K.-S. Choi, J. Am. Chem. Soc., 2012, 134, 2186–2192 CrossRef CAS PubMed.
- F. F. Abdi and R. van de Krol, J. Phys. Chem. C, 2012, 116, 9398–9404 CAS.
- K. J. McDonald and K.-S. Choi, Energy Environ. Sci., 2012, 5, 8553–8557 CAS.
- C. Ding, J. Shi, D. Wang, Z. Wang, N. Wang, G. Liu, F. Xiong and C. Li, Phys. Chem. Chem. Phys., 2013, 15, 4589–4595 RSC.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 Search PubMed.
- F. Le Formal, K. Sivula and M. Grätzel, J. Phys. Chem. C, 2012, 116, 26707–26720 CAS.
- L. Peter, J. Solid State Electrochem., 2013, 17, 315–326 CrossRef CAS PubMed.
- S. R. Pendlebury, M. Barroso, A. J. Cowan, K. Sivula, J. W. Tang, M. Gratzel, D. Klug and J. R. Durrant, Chem. Commun., 2011, 47, 716–718 RSC.
- A. J. Cowan, J. W. Tang, W. H. Leng, J. R. Durrant and D. R. Klug, J. Phys. Chem. C, 2010, 114, 4208–4214 CAS.
- N. Aiga, Q. Jia, K. Watanabe, A. Kudo, T. Sugimoto and Y. Matsumoto, J. Phys. Chem. C, 2013, 117, 9881–9886 CAS.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef CAS PubMed.
- F. M. Pesci, G. Wang, D. R. Klug, Y. Li and A. J. Cowan, J. Phys. Chem. C, 2013, 117, 25837–25844 CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- F. M. Pesci, A. J. Cowan, B. D. Alexander, J. R. Durrant and D. R. Klug, J. Phys. Chem. Lett., 2011, 2, 1900–1903 CrossRef CAS.
- Z. Huang, Y. Lin, X. Xiang, W. Rodriguez-Cordoba, K. J. McDonald, K. S. Hagen, K.-S. Choi, B. S. Brunschwig, D. G. Musaev, C. L. Hill, D. Wang and T. Lian, Energy Environ. Sci., 2012, 5, 8923–8926 CAS.
- K. Sayama, A. Nomura, T. Arai, T. Sugita, R. Abe, M. Yanagida, T. Oi, Y. Iwasaki, Y. Abe and H. Sugihara, J. Phys. Chem. B, 2006, 110, 11352–11360 CrossRef CAS PubMed.
- Z. B. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res., 2010, 25, 3–16 CrossRef CAS.
- M. Valant and D. Suvorov, J. Am. Ceram. Soc., 2000, 83, 2721–2729 CrossRef CAS PubMed.
- J. J. Kelly and D. Vanmaekelbergh, Electrochim. Acta, 1998, 43, 2773–2780 CrossRef CAS.
- A. Reynal, F. Lakadamyali, M. A. Gross, E. Reisner and J. R. Durrant, Energy Environ. Sci., 2013, 6, 3291–3300 CAS.
- We are aware that a lower donor density (1015 cm−3) was reported with another technique (Hall effect measurements) in ref. 5, which yields a significantly wider width of the space charge layer than 90 nm reported herein.
- Photoelectrochemical Hydrogen Production, ed. R. van de Krol and M. Grätzel, Springer, US, 2012 Search PubMed.
- J. W. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- S. R. Pendlebury, A. J. Cowan, M. Barroso, K. Sivula, J. Ye, M. Gratzel, D. R. Klug, J. Tang and J. R. Durrant, Energy Environ. Sci., 2012, 5, 6304–6312 CAS.
- K. P. S. Parmar, H. J. Kang, A. Bist, P. Dua, J. S. Jang and J. S. Lee, ChemSusChem, 2012, 5, 1926–1934 CrossRef CAS PubMed.
- A. J. Cowan, C. J. Barnett, S. R. Pendlebury, M. Barroso, K. Sivula, M. Gratzel, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2011, 133, 10134–10140 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images (top and cross sectional views), XRD pattern of the undoped BiVO4 photoanode, IPCE and UV-vis spectra, Mott–Schottky plot, transient absorption spectra under applied bias, time constants related to back electron/hole recombination and water oxidation, fast phase recombination amplitude as a function of applied bias, comparison of back electron/hole recombination measured with two techniques, fit parameters of transient absorption decays. See DOI: 10.1039/c4sc00469h |
| This journal is © The Royal Society of Chemistry 2014 |