 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDicyanopyrazine-derived push–pull chromophores for highly efficient photoredox catalysis†
Yu Zhao‡
a,
Chenhao Zhang‡a,
Kek Foo Chinc,
Oldřich Pytelab,
Guo Weia,
Hongjun Liua,
Filip Bureš*b and
Zhiyong Jiang*a
aKey Laboratory of Natural Medicine and Immuno-Engineering of Henan Province, Henan University, Kaifeng, Henan 475004, P. R. China. E-mail: chmjzy@henu.edu.cn; filip.bures@upce.cz
bInstitute of Organic Chemistry and Technology, University of Pardubice, Faculty of Chemical Technology, Studentská 573, Pardubice, 53210, Czech Republic
cDivision of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore
First published on 30th June 2014
Abstract
Here, we report dicyanopyrazine (DPZ)-derived push–pull chromophores, easily prepared and tunable organic compounds, as new kinds of photoredox catalysts. In particular, the DPZ derivative H, containing 2-methoxythienyl as electron-donating moiety, exhibits a broad absorption of visible light with an absorption edge up to 500 nm and excellent redox properties, and has been demonstrated as a desirably active and efficient photoredox catalyst in four challenging kinds of photoredox reactions. The amount of catalyst in most reactions is less than 0.1 mol% and even 0.01 mol%, representing the lowest catalyst loading in the current photoredox organocatalysis.
Introduction
Towards designing new reactions, mild and green conditions are always favoured to minimize energy consumption on the standpoint of green chemistry.1 In this context, photoredox catalysis (photo-induced single electron transfer, SET), which utilizes visible light as a naturally abundant and clean energy source and does not need any special instrumentation nor apparatus, has attracted considerable attention from synthetic organic chemists.2–8 In particular, the visible light absorbing photoredox catalysts, which are able to transform solar to chemical energy, play the most important role in photocatalytic reactions.In recent years, various readily available compounds, such as polypyridyl complexes of ruthenium9,10 and iridium11,12 as well as organic dyes,13,14 have been utilized as the photoredox catalysts. Other designed photocatalysts with tailored structures include platinum(II),15 gold(III),16 palladium(II)17 and copper(II)18,19 complexes and acridinium ions20,21 (Fig. 1). Obviously, the photoredox catalyst simultaneously equipped with the following features still remains highly desirable: (1) a readily available/prepared organic compound. The organic compound should more likely reduce the cost and the potential contamination of target products with toxic heavy metals;14 (2) tunable structure. The easily modified structure of photocatalyst will benefit to optimize the photophysical properties, such as the absorption edge and intensity in the visible range as well as the redox potentials. In comparison with the well-established metal complexes, the organic photoredox catalyst with tunable structure is still deficient;20–22 (3) high activity and efficiency. A universal substance able to catalyse a variety of challenging and unprecedented photoredox reactions is always desirable, which is determined by the activity of photocatalyst. Meanwhile, the amount of the catalyst loaded in the reaction is always an important index to evaluate its efficiency. To the best of our knowledge, few examples23–25 with less than 0.1 mol% of the catalyst were presented to date unless using high power light sources17,26 or in flow.27–29 Most important, the example of photoredox reaction with less than 0.1 mol% organic catalyst is not reported. In principle, the activity and efficiency of photocatalyst could be improved by optimizing its photophysical properties.
 | ||
| Fig. 1 Selected examples of photoredox catalysts reported to date. | ||
In 2012, push–pull molecules based on 5,6-disubstituted pyrazine-2,3-dicarbonitrile (dicyanopyrazine, DPZ) were presented and their nonlinear optical properties were investigated by our group (e.g. DPZ derivatives A–B, Fig. 2).30 These organic compounds can be readily prepared from available starting materials such as diaminomaleodinitrile (DAMN) in excellent yields and represent stable D–π–A chromophores with intramolecular charge-transfer (ICT), which resembles the MLCT (metal to ligand charge transfer) in aforementioned metal complexes. Furthermore, their redox potentials are similar to known photoredox catalysts (e.g. E1/2(red) of eosin Y: −1.0 V, rhodamine B: −0.8 V, A: −1.24 V, B: −0.87 V vs. saturated calomel electrode [SCE]) while the absorption maxima locate at the visible light area (λmax of A: 471 nm, B: 499 nm). Most importantly, their optical and electrochemical properties can readily be tuned by the variation of the appended electron-donors. Based on these features, we envisioned that DPZ derivatives could probably serve as aforementioned photoredox catalysts.
 | ||
| Fig. 2 The structures and convenient/improved preparations of DPZ derivatives A–H. | ||
Results and discussion
Preparation and properties of DPZ
In this respect, DPZ derivatives C–H were designed and synthesized, possessing either electron withdrawing or donating (hetero)aromates at the pyrazine C5/C6 positions such as in C–D or E–H (Fig. 2). The method used for the construction of dicyanopyrazine moiety involves condensation of DAMN with 1,2-dicarbonyl compounds (benzils), affording DPZ derivatives (C–G) in 89–92% yields. However, 5-methoxythienyl, as the most electron-rich moiety, made this reaction sluggish and only 3% yield of H was obtained. Our previous findings30 prompted us to attempt twofold cross-coupling reaction of 5,6-dichloropyrazine-2,3-dicarbonitrile with 5-methoxythiophen-2-ylboronic acid pinacol ester (Fig. 2). Under the optimized conditions (see the ESI†), this reaction afforded the desired product H in 83% yield. Notably, the preparation of H in a gram scale could conveniently be carried out via this efficient protocol (Fig. 2).The properties of DPZ derivatives C–H were further studied by electrochemical measurements, absorption spectra and DFT calculations (Table 1, Fig. 3 and ESI†). The HOMO–LUMO energies, their differences and the position of the longest-wavelength absorption maxima can be considered as the most important electronic parameters. The first oxidation potential respective the HOMO energy decrease with increasing electron donating ability of the substituents and thus enhanced ICT. As a result, the electrochemical gap (Eg) decreases from 3.70 to 2.82 eV. Likewise, the longest-wavelength absorption maxima λmax shift bathochromically (Fig. 3) and the respective optical as well as the calculated gaps EDFTg decrease significantly. It is noteworthy, that electrochemical, optical and calculated gaps show the same trend and correlate very tightly (see ESI†). Hence, we can consider both experiments and DFT calculation as reliable tools describing the electronic parameters of C–H. As can be seen, the electronic parameters of DPZ derivative H are exceptional. In contrast to G (Eg = 3.27 eV; λmax = 391 nm (3.17 eV), EDFTg = 3.37 eV, μ = 12.88 D), two additional methoxy substituents in H lowered the electrochemical/calculated gap by 0.45/0.39 eV, shifted the λmax bathochromically by 57 nm and increased the molar absorption coefficient as well as the ground state dipole moment. Obviously, the electronic properties of DPZ derivative H (Eg = 2.82 eV, λmax = 448 nm (2.78 eV), ε = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 mol−1 dm3 cm−1, EDFTg = 2.97 eV, μ = 18.26 D) are well tuned and suited for the photoredox catalysis (e.g. Ru(bpy)3Cl2, λmax = 452 nm, Eg = 2.62 eV). As expected, the HOMO and the LUMO in H are clearly separated and localized on the donor and acceptor parts of the molecule (Fig. 2, for C–G see ESI†).
500 mol−1 dm3 cm−1, EDFTg = 2.97 eV, μ = 18.26 D) are well tuned and suited for the photoredox catalysis (e.g. Ru(bpy)3Cl2, λmax = 452 nm, Eg = 2.62 eV). As expected, the HOMO and the LUMO in H are clearly separated and localized on the donor and acceptor parts of the molecule (Fig. 2, for C–G see ESI†).
| DPZ | E1/2(ox1)a [V] | E1/2(red1)a [V] | EHOMOb [eV] | ELUMOb [eV] | Egc [eV] | λmax [nm (eV)]/ε × 10−3d [mol−1 dm3 cm−1] | EDFTge [eV] | μe [D] |
|---|---|---|---|---|---|---|---|---|
| a E1/2(ox1)/E1/2(red1) are half-wave potentials of the first oxidation/reduction measured in DCM versus SCE.b EHOMO/LUMO = E1/2(ox1/red1) + 4.80.31c EHOMO–ELUMO electrochemical gap.d Longest-wavelength absorption maxima (optical gap)/molar absorption coefficient (DCM).e DFT calculated HOMO–LUMO gap and ground state dipole moment (B3LYP/6-311++G(2df,p)//B3LYP/6-311++G(2df,p), scrf = (solvent = dichloromethane)). | ||||||||
| C | +2.30 | −1.40 | −7.10 | −3.41 | 3.70 | 340(3.65)/15.3 | 3.93 | 11.38 |
| D | +2.27 | −1.41 | −7.07 | −3.39 | 3.68 | 345(3.59)/13.2 | 3.92 | 7.43 |
| E | +1.90 | −1.44 | −6.70 | −3.36 | 3.34 | 383(3.24)/19.7 | 3.58 | 10.80 |
| F | +1.95 | −1.36 | −6.75 | −3.44 | 3.31 | 379(3.27)/17.4 | 3.54 | 12.69 |
| G | +1.92 | −1.35 | −6.72 | −3.45 | 3.27 | 391(3.17)/14.6 | 3.37 | 12.88 |
| H | +1.37 | −1.45 | −6.17 | −3.35 | 2.82 | 448(2.78)/21.5 | 2.97 | 18.26 |
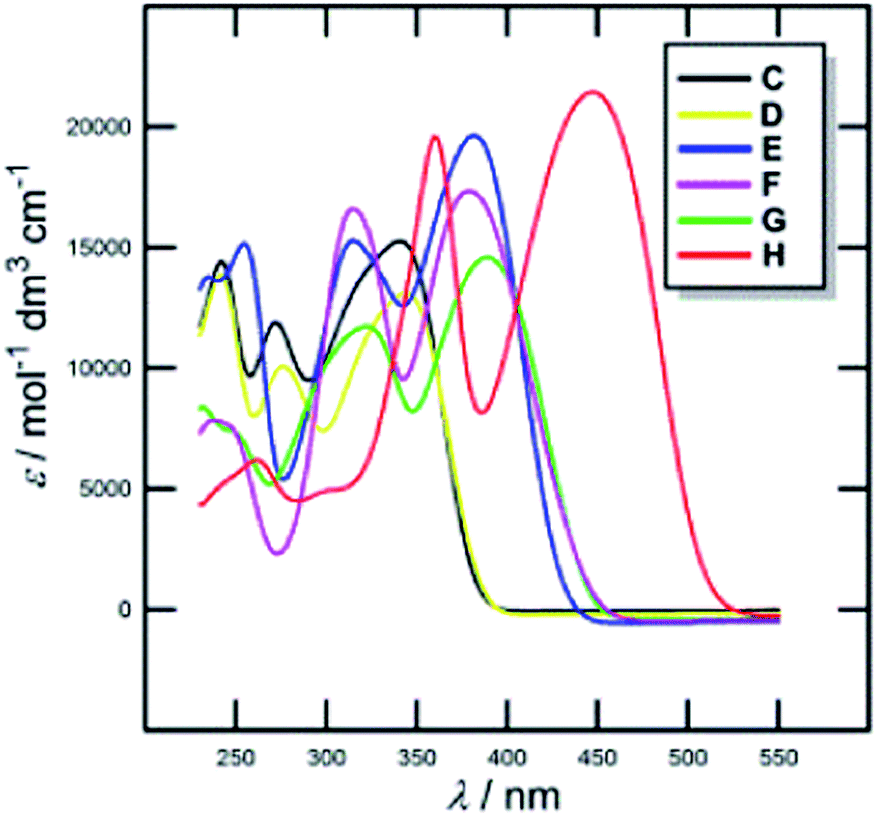 | ||
| Fig. 3 UV-Vis absorption spectra of DPZ derivatives C–H in DCM (c = 2 × 10−5 M). | ||
DPZ derivatives in cross-dehydrogenative-coupling reactions
Since introduced by Stephenson and co-workers in 2010,32 the visible light irradiated cross-dehydrogenative-coupling (CDC) reaction of N-aryl-tetrahydroisoquinolines and nitroalkanes has become the most favorable protocol to evaluate the efficiency of catalysts.15,17,27,33–41 To probe the feasibility of DPZ derivatives in photoredox catalysis, we initially performed the CDC reaction of N-phenyl-tetrahydroisoquinoline 1a and nitromethane 2a in the presence of 2.0 mol% A–H at 28 °C in ambient air and irradiated by a 9 W fluorescent lamp (Table 2, entries 1–8). All DPZ derivatives A–H could catalyse the reaction while H showed an outstanding activity to access the desired adduct 3a with 96% conversion in 2 hours (Table 2, entry 8). Obviously, the reaction results depend on the efficiency of the ICT in the used catalyst. Reduced HOMO–LUMO gap, bathochromically shifted λmax and enhanced absorption intensity of the catalysts resulted in an increased conversion, which is being superior with catalyst H. The reaction could be finished with complete conversion about 5 hours even if the amount of H was decreased to 0.1 mol% (Table 2, entry 9). DMF as solvent cleanly provided the desired product 3a with the same reactivity (Table 2, entry 10).
|
|||
|---|---|---|---|
| Entry | DPZ catalyst (mol%) | T [h] | Conversionb [%] |
| a The reactions were performed with 0.05 mmol of 1a in 0.5 mL of 2a in the presence of catalyst at 28 °C irradiated by a 9 W household fluorescent lamp.b Determined by 1H NMR of crude reaction mixture.c 0.05 mmol of 1a and 0.5 mmol of 2a in 0.5 mL DMF under the given reaction conditions. | |||
| 1 | A (2.0) | 2 | 12 |
| 2 | B (2.0) | 2 | 7 |
| 3 | C (2.0) | 2 | 10 |
| 4 | D (2.0) | 2 | 8 |
| 5 | E (2.0) | 2 | 38 |
| 6 | F (2.0) | 2 | 49 |
| 7 | G (2.0) | 2 | 76 |
| 8 | H (2.0) | 2 | 96 |
| 9 | H (0.1) | 5 | 100 |
| 10c | H (0.1) | 5 | 100 |

The reactions of a variety of N-aryltetrahydroisoquinolines 1 with nitromethane 2a as solvent were subsequently attempted under established conditions, providing the corresponding adducts 3a–g in 87–95% yields within 5 hours (Table 3, entries 1–7). Importantly, 85% yield of 3a was attained within 24 hours even when 0.01 mol% of H was used under the irradiation with 9 W fluorescent lamp. The reaction seems to be faster when irradiated by sunlight [Table 3, entry 1, see items (iii) and (iv) in footnote (b)]. The reactions of 1a with other nitroalkanes (2b–c) gave adducts 3h–i in excellent yields after 5 hours (Table 3, entries 8 and 9).
| a Detailed conditions for these reactions are described in the ESI.†b (i) 0.05 mol% of H was used, 9 W fluorescent lamp, 28 °C, t = 12 h, yield = 86%; (ii) 0.01 mol% of H was used, 9 W fluorescent lamp, 28 °C, t = 24 h, yield = 85%; (iii) 0.05 mol% of H was used, sunlight (30 °C), t = 3.5 h, yield = 83%; (iv) 0.01 mol% of H was used, sunlight (30 °C), t = 3.5 h, yield = 60%.c 0.15 mmol scale, DMF as solvent. |
|---|
 |
Next, we screened a series of CDC reactions between different tertiary/secondary amines and various nucleophiles under the established conditions (28 °C, DMF as solvent, ambient air and irradiation with 9 W fluorescent lamp, Table 3, entries 10–15). The reaction27,35,36,41–43 of N-phenyl-tetrahydroisoquinoline 1a and TMSCN 2d was firstly processed in the presence of 0.1 mol% of H, and the oxidative Strecker adduct 3j was obtained within 2 hours in 87% yield (Table 3, entry 10). With the same catalyst loading and the help of pyrrolidine/TFA, the powerful catalytic ability of H was further demonstrated by the reaction16,33,36,38,40,44 of 1a and acetone 2e, affording Mannich adduct 3k in 93% yield after 4 hours (Table 3, entry 11). To introduce a challenging hetero-quaternary stereogenic center adjacent to N-aryl-tetrahydroisoquinolines, we attempted the first example of oxidative Mannich reaction of 1a and 5H-oxazol-4-one 2f via photoredox catalysis (Table 3, entry 12). It was discovered that, in the presence of only 0.01 mol% of H, the desired adduct 3l was obtained in 82% yield with 10:1 dr after 4 hours. Use of 0.01 mol% of H as catalyst also resulted in the complete reaction34,36,45 between 1a and diethyl phosphonate 2g, to furnish the P–C bond formation and afford the product 3m in 81% yield within 7 hours (Table 3, entry 13).
Over the past few years, the CDC reaction32,34,40 of N-arylpyrrolidines and nitromethane has been attempted in photoredox catalysis under different conditions; however, the reported reactions were always sluggish with poor yields, and N-arylpyrrolidines were thus recognized as the non-activated amines. Hence, we were intrigued to conduct the formidable reaction of N-(4-tert-butylphenyl)pyrrolidine 4 and nitromethane 1 (Table 3, entry 14). We were pleased that the desired adduct 5 could be obtained in 76% yield after 45 hours when 2.0 mol% of H was used. Due to the longest-wavelength absorption maxima of H reaching blue light area, the reaction was also tested under the irradiation of blue Light Emitting Diode (LED, 4 W, house-hold), and completed in 15 hours to deliver adduct 5 in 80% yield. On the other hand, linear secondary amines exhibit very poor activity in photocatalytic CDC reaction, and the singlet oxygen protocol combined with a special photoreactor seems necessary.29 Accordingly, we turned our attention to the reaction of dibenzylamine 6 and TMSCN 2d (Table 3, entry 15). It was found that the reaction could be finished smoothly in 17 hours by using 0.1 mol% of H, to afford the adduct 7 in 84% yield.
Mechanism of the photochemical CDC reactions
We were also interested in fundamental principles of DPZ action in photoredox catalyzed reactions. In the above CDC reactions, H2O2 can be envisaged as a reasonable by-product. As Fig. 4 shown, hydrogen peroxide could clearly be detected in the crude reaction mixture of 1a and 2a catalysed by H. The presence of H2O2 was confirmed by the iodide test (KI in glacial acetic acid), in which a colour change to dark red was detected. | ||
| Fig. 4 Test for the production of hydrogen peroxide in the reaction mixture of 1a and 2a. (a) KI and starch in glacial acetic acid, colourless. (b) The reaction mixture of 1a and 2a after reaction completed, brown. (c) Addition of solution (b) to solution (a), dark red. (d) Addition of 30% of H2O2 in solution (a). KI (0.2 M), aqueous acetic acid (0.2 M), starch (4 mg mL−1), DPZ H (0.1 μmol, 14.14 μL, 2.5 mg mL−1 in CH3NO2), 1a (0.1 mmol), CH3NO2 (1.0 mL), reaction time: 5 h, 28 °C. | ||
Considering three plausible pathways, such as singlet-oxygen oxidation, photoredox reaction or both of these two reactions, we subsequently conducted the study on solvent effects since the lifetime of singlet oxygen highly depends on the solvent used. The singlet oxygen lifetime should be significantly longer in deuterated solvents in comparison to their protonated counterparts.46–48 Therefore, a reaction of 1a and 2a was performed in the presence of 0.1 mol% of DPZ derivative H at 25 °C in both CH3CN and CD3CN solvent. The results of the kinetic measurements of the product conversions are indicated in Fig. 5. From these dependencies we can conclude that singlet oxygen was likely not a key participant in the reaction as no significant increase in the reaction rate was observed in CD3CN. Mechanistically, we suggest a plausible mechanism on the above CDC reactions with DPZ derivative H as catalyst (Fig. 6), which is a photoredox protocol as Stephenson previously disclosed for the ruthenium or iridium complexes.32 This reaction mechanism involves excitation of the DPZ H catalyst by visible light, reductive quench (single-electron transfer, SET) from the amino-substrate to form radical cation/anion R3N˙+ and DPZ H˙−. Subsequent reaction with the nitroalkane or eventually oxygen ([O]) regenerates the H-catalyst and formed [O]˙− deprotonates the trialkylammonium radical cation R3N˙+ to iminium salt, which undergoes nucleophilic attack of the nucleophile used.
 | ||
Fig. 5 Photocatalytic CDC reaction of 1a and 2a (or 2a-d3) to give adduct 3a in the presence of DPZ derivative H; reaction kinetics was monitored by using 1H NMR spectroscopy; CH3NO2 (■), CD3NO2 ( ); DPZ H (0.05 μmol, 7.0 μL, 2.5 mg mL−1), 1a (0.5 mmol), CH3NO2 or CD3NO2 (0.5 mL), reaction time: 6 h, 25 °C. ); DPZ H (0.05 μmol, 7.0 μL, 2.5 mg mL−1), 1a (0.5 mmol), CH3NO2 or CD3NO2 (0.5 mL), reaction time: 6 h, 25 °C. | ||
 | ||
| Fig. 6 Plausible mechanism of CDC reaction catalyzed by DPZ H. | ||
On the basis of the above observations and the proposed mechanism, we can deduce the following fundamental structure–catalytic activity relationships:
• Bathochromically shifted absorption maxima (λmax), high molar absorption coefficient (ε) and small HOMO–LUMO gap (Eg) of the catalyst improve its excitation ability by low-energy visible light (compare for H and C–G, Table 1).
• Principal changes in the properties of DPZ derivatives C–H are caused by the donor-centered HOMO (EHOMO range from −7.10 to −6.17 eV, Table 1).
• The LUMO localized on the dicyanopyrazine acceptor remained almost unaltered in all derivatives (ELUMO range from −3.45 to −3.39 eV, Table 1).
• Reduced HOMO–LUMO gap of the catalyst facilitates the generation of radicals (SET, Fig. 6).
• The conversion of CDC reaction depends linearly and tightly on the electrochemical as well as optical HOMO–LUMO gaps of the used X-shaped push–pull DPZs (see the correlation in Fig. S12 in the ESI†).
• Planar and polariable π-system of the catalyst help to stabilize the radical anion e.g. DPZ H˙− (Fig. 6) through delocalization.
• Well-tuned ICT helps to reduce HOMO–LUMO gap (see charge separation in H, Fig. 2) but does not cause high electron saturation of the cyano acceptors.
• Higher electron saturation of the cyano groups would have detrimental effect on the formation of the radical anion e.g. DPZ H˙− (Fig. 6).
• These suppositions can be demonstrated as follows: DPZ derivatives bearing strong electron NMe2 donors A (Eg = 2.26 eV) and B (Eg = 1.91 eV) showed lower Eg values and more bathochromically shifted absorption maxima than H, but their catalytic activity is very low (Table 2). This is most likely caused by high D–A coupling between NMe2 and CN groups and thus higher contribution of the quinoid/zwitterionic form with negatively charged cyano groups (see Fig. S13 in the ESI†).
H catalyses other challenging reactions
To further evaluate the performance of DPZ derivative H, other challenging or unprecedented photoredox reactions were successively attempted (Fig. 7). We firstly carried out an oxidation of N-methyl-2-(phenylamino)acetamide 8 using 1.0 mol% of H in DMF at 28 °C under oxygen atmosphere and irradiation by a 9 W fluorescent lamp (eqn (1)). We found that the reaction was finished within 7 hours to give unsymmetric oxalamide 9. Indeed, despite the well-established photocatalytic oxidation of tertiary amines,49,50 the transformation of activated secondary amine to amide has not been reported yet.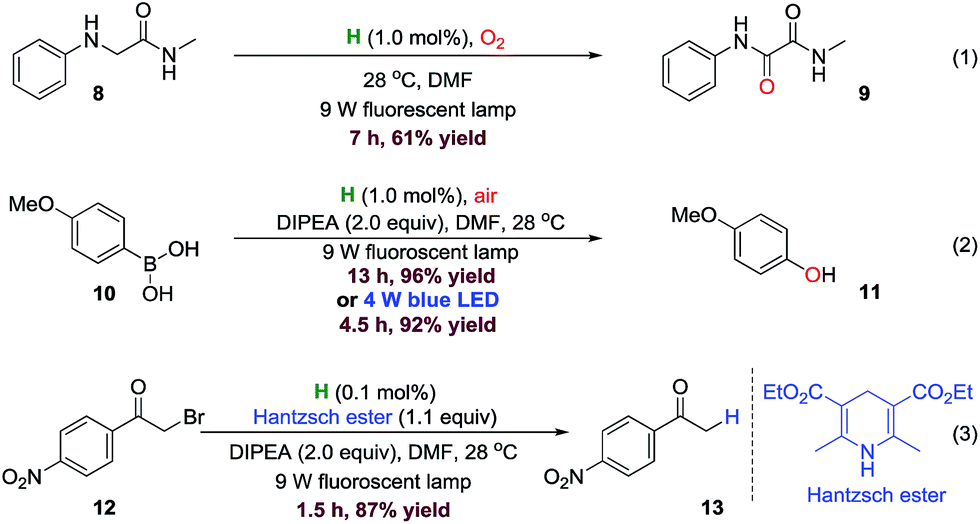 | ||
| Fig. 7 Photocatalytic oxidation, oxidative hydroxylation and reductive dehalogenation. | ||
In 2012, Jørgensen and Xiao and co-workers introduced the first oxidative hydroxylation of aryl boronic acids by utilizing 2.0 mol% of Ru complex as catalyst, in which 36 W fluorescence lamp was used as light source.51 Moreover, Acid Red 87, the organic compound, has also been demonstrated as the photoredox catalyst, but the reaction became slower.51 Scaiano group52 gave a metal-free example of oxidative hydroxylation of aryl boronic acids with methylene blue as catalyst; the catalyst amount could be decreased to 1.0 mol% and the reactions became faster, but two 90 W warm white LEDs as the light source and pure oxygen atmosphere were necessary. Therefore, we investigated this demanding reaction with the DPZ derivative H as catalyst. As eqn (2) shown (Fig. 7), the oxidative hydroxylation of 4-methoxy-phenylboronic acid 10 was performed with 1.0 mol% of H in ambient air and irradiated by a 9 W fluorescent lamp, and the desired product 11 was obtained in 96% yield after 13 hours. The reaction could be accelerated by utilizing 4 W LEDs as the light source, in which the product 11 could be obtained in 92% yield within 4.5 hours.
We subsequently explored the reductive dehalogenation of α-bromoacetophenones. In comparison with the excellent work by Zeitler and co-workers [2.5 mol% eosin Y, 1 W high-power LEDs (λ = 530 nm), 18 h, 83% yield],14 the reductive dehalogenation of bromoacetophenone 12 could be finished within 1.5 hours in the presence of 0.1 mol% of H under the irradiation by a 9 W house-hold fluorescent lamp, to access the product 13 in 87% yield (Fig. 7, eqn (3)).
Conclusion
Dicyanopyrazine-derived (DPZ) push–pull chromophores have been demonstrated as a new kind of organic catalysts for photoredox catalysis. 2-Methoxythienyl as an electron-donating moiety provides a DPZ derivative (H) with a broad absorption edge reaching up to 500 nm as well as the excellent redox properties. Moreover, all the aforementioned features are well balanced and enable the DPZ derivative H to become the desirably active and efficient photoredox catalyst, which was demonstrated by more than four kinds of photocatalytic organic reactions. Without the aid of high power light sources or in flow, the amount of catalyst in most reactions is always less than 0.1 mol% and even 0.01 mol%, which represents the lowest catalyst loading in the current photoredox organocatalysis. Other excellent performances on the DPZ derivative H-catalyzed reactions include lower power light source, air atmosphere while short reaction time and higher yields. Further investigations, which involve the use of DPZ derivatives as catalyst in novel photocatalytic reactions, are ongoing in our labs and will be reported in due course.Acknowledgements
This research was supported by the Czech Science Foundation (13-01061S), NSFC (nos. 21072044, U1204207) and the Program for New Century Excellent Talents in University of Ministry of Education (NCET-11-0938).Notes and references
- P. Tundo, P. Anastas, D. S. Black, J. Breen, T. Collins, S. Memoli, J. Miyamoto, M. Polyakoff and W. Tumas, Pure Appl. Chem., 2009, 72, 1207 Search PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- M. A. Ischay and T. P. Yoon, Eur. J. Org. Chem., 2012, 3359 CrossRef CAS.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed.
- H. Liu, W. Feng, C. W. Kee, Y. Zhao, D. Leow, Y. Pan and C.-H. Tan, Green Chem., 2010, 12, 953 RSC.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed.
- J.-J. Zhong, Q.-Y. Meng, G.-X. Wang, Q. Liu, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2013, 19, 6443 CrossRef CAS PubMed.
- Q. Xue, J. Xie, H. Jin, Y. Cheng and C. Zhu, Org. Biomol. Chem., 2013, 11, 1606 CAS.
- W.-P. To, Y. Liu, T.-C. Lau and C.-M. Che, Chem.–Eur. J., 2013, 19, 5654 CrossRef CAS PubMed.
- J.-M. Kern and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1987, 546 RSC.
- M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem.–Eur. J., 2012, 18, 7336 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed.
- T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588 CrossRef CAS PubMed.
- S. Guo, H. Zhang, L. Huang, Z. Guo, G. Xiong and J. Zhao, Chem. Commun., 2013, 49, 8689 RSC.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed.
- M. A. Ischay, M. S. Ament and T. P. Yoon, Chem. Sci., 2012, 3, 2807 RSC.
- S. Samanta, S. Das and P. Biswas, J. Org. Chem., 2013, 78, 11184 CrossRef CAS PubMed.
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144 CrossRef CAS PubMed.
- M. Neumann and K. Zeitler, Org. Lett., 2012, 14, 2658 CrossRef CAS PubMed.
- D. B. Ushakov, K. Gilmore, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 53, 557 CrossRef CAS PubMed.
- F. Bureš, H. Čermáková, J. Kulhánek, M. Ludwig, W. Kuznik, I. V. Kityk, T. Mikysek and A. Růžička, Eur. J. Org. Chem., 2012, 529 CrossRef.
- B. W. D'Andrade, S. Datta, S. R. Forrest, P. Djurovich, E. Polikarpov and M. E. Thompson, Org. Electron., 2005, 6, 11 CrossRef PubMed.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS PubMed.
- Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682 RSC.
- D. P. Hari and B. König, Org. Lett., 2011, 13, 3852 CrossRef CAS PubMed.
- Z. Xie, C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056 CrossRef CAS PubMed.
- M. Rueping, J. Zoller, D. C. Fabry, K. Poscharny, R. M. Koenigs, T. E. Weirich and J. Mayer, Chem.–Eur. J., 2012, 18, 3478 CrossRef CAS PubMed.
- J.-L. Wang, C. Wang, K. E. deKrafft and W. Lin, ACS Catal., 2012, 2, 417 CrossRef CAS.
- Q. Liu, Y.-N. Li, H.-H. Zhang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2012, 18, 620 CrossRef CAS PubMed.
- M. Neumann and K. Zeitler, Org. Lett., 2012, 14, 2658 CrossRef CAS PubMed.
- L. Möhlmann, M. Baar, J. Rieβ, M. Antonietti, X. Wang and S. Blechert, Adv. Synth. Catal., 2012, 354, 1909 CrossRef.
- M. Rueping, C. Vila and T. Bootwicha, ACS Catal., 2013, 3, 1676 CrossRef CAS.
- Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341 RSC.
- M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 12709 RSC.
- M. Rueping, C. Vila, R. M. Koenigs, K. Poscharny and D. C. Fabry, Chem. Commun., 2011, 47, 2360 RSC.
- M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 8679 RSC.
- J. R. Hurst, J. D. McDonald and G. B. Schuster, J. Am. Chem. Soc., 1982, 104, 2065 CrossRef CAS.
- P. R. Ogilby and C. S. Foote, J. Am. Chem. Soc., 1983, 105, 3423 CrossRef CAS.
- A. G. Griesbeck, V. Schlundt and J. M. Neudörfl, RSC Adv., 2013, 3, 7265 RSC.
- P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672 CrossRef CAS PubMed.
- J. H. Park, K. C. Ko, E. Kim, N. Park, J. H. Ko, D. H. Ryu, T. K. Ahn, J. Y. Lee and S. U. Son, Org. Lett., 2012, 14, 5502 CrossRef CAS PubMed.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef CAS PubMed.
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures on the preparation of DPZ derivatives, quantum chemical calculations, procedures for photoredox catalysed reactions, characterization data of data and NMR spectra. See DOI: 10.1039/c4ra05525j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |