
Improved hydrogen production from formic acid under ambient conditions using a PdAu catalyst on a graphene nanosheets–carbon black support†
Abstract
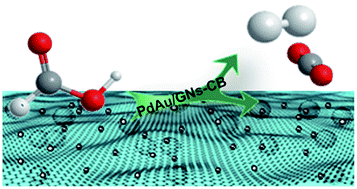
Formic acid (FA) has great potential as a suitable liquid source for hydrogen and hydrogen storage materials, provided highly active and selective dehydrogenation catalysts under ambient conditions are developed. Here, well-dispersed bimetallic gold–palladium (PdAu) nanoparticles (NPs) grown on graphene nanosheets–carbon black (GNs–CB) composite supports are synthesized via a facile co-reduction method, wherein the GNs–CB composite support proved to be a powerful dispersion agent and a distinct support for the PdAu NPs. Interestingly, the resultant PdAu/GNs–CB catalyst manifests high selectivity and exceedingly high activity to complete the decomposition of FA at room temperature.
 Please wait while we load your content...
Please wait while we load your content...

