Intramolecular oxidative cyclization of alkenes by rhodium/cobalt porphyrins in water†
Lin
Yun
,
Zikuan
Wang
and
Xuefeng
Fu
*
Beijing National Laboratory for Molecular Sciences, State Key Lab of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China. E-mail: fuxf@pku.edu.cn; Tel: +86 10 6275 6035
First published on 13th June 2014
Abstract
Intramolecular oxidative cyclization of alkenes provides a unique pathway to obtain unsaturated heterocyclic compounds. The rhodium(III)/cobalt(III) tetra(p-sulfonatophenyl)porphyrin ((TSPP)MIII) in water was observed to mediate the intramolecular oxidative functionalization of alkenes to a series of unsaturated oxygen heterocyclic compounds.
The activation of alkenes by coordination to metal centers and subsequent functionalization is one of the most widely exploited synthetic methodologies in the functionalization of organic molecules.1–3 Intramolecular oxidative cyclization of alkenes provides a unique pathway to obtain unsaturated heterocyclic compounds. Transition metal complex mediated intramolecular Wacker-type oxidative alkoxylation provides a straightforward access to oxygen heterocycles.4 The retained C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds allow further functionalization of the resulting heterocycles. However, this process is far less developed compared with intramolecular hydrofunctionalization of alkenes5–7 and most oxidative cyclization/functionalization studies focused on palladium catalysis.1,8 Additionally, the importance of alkene transformations and objectives of green chemistry justify the continuing search for new classes of catalyst materials and new pathways leading to novel oxidative functionalization in water.
C double bonds allow further functionalization of the resulting heterocycles. However, this process is far less developed compared with intramolecular hydrofunctionalization of alkenes5–7 and most oxidative cyclization/functionalization studies focused on palladium catalysis.1,8 Additionally, the importance of alkene transformations and objectives of green chemistry justify the continuing search for new classes of catalyst materials and new pathways leading to novel oxidative functionalization in water.
Rhodium porphyrin complexes demonstrate a wide range of important substrate reactions.9,10 Groves demonstrated intramolecular anti-Markovnikov hydrofunctionalization of olefins mediated by the tetraphenyl porphyrin rhodium hydride complex (Scheme 1A).10 Previous work from our group has accomplished stoichiometric aerobic oxidation of olefins in water mediated by a water soluble rhodium porphyrin complex (TSPP)RhIII.11 Rhodium porphyrin β-hydroxyalkyl complexes were observed and isolated as intermediates, which underwent β-hydrogen elimination (BHE) to give methyl ketones as the oxidation products (Scheme 1B). Based on these studies, we extended our interest towards the intramolecular oxidative cyclization of alkenes to form unsaturated heterocycles (Scheme 1C) through a four-step cycle involving: (1) alkene coordination to (TSPP)RhIII; (2) intramolecular nucleophilic attack by the –OH group towards activated alkenes; (3) hydrogen elimination to give unsaturated cyclization products and (4) the reduced metal complex (TSPP)RhI was aerobically oxidized to (TSPP)RhIII.
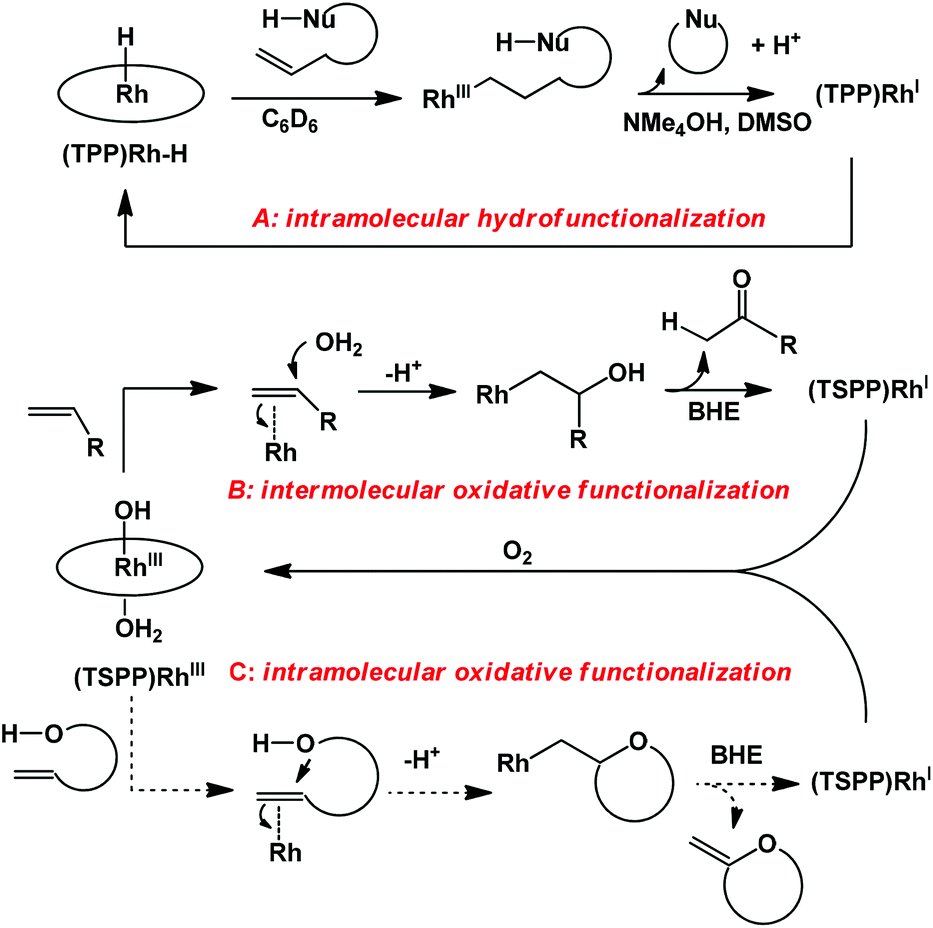 | ||
| Scheme 1 The rhodium porphyrin complex mediated transformation of alkenes. | ||
Intramolecular nucleophilic activation of alkenes
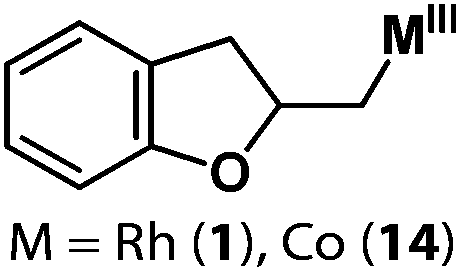
Allylphenol was selected as the model substrate which showed good reactivity as previously reported.8,12 Addition of 2-allylphenol to a borate buffered (pH = 8.0) D2O solution of (TSPP)RhIII at room temperature resulted in an immediate color change from sanguine to orange-red. The 1H NMR experiment revealed complete conversion of (TSPP)RhIII to the alkyl rhodium complex 1 which was conveniently identified by the appearance of a set of high field resonances attributed to the (2,3-dihydrobenzofuran-2-yl)methyl group bonded to the rhodium center. The 1H NMR resonances centered at −5.67 and −5.81 ppm were associated with the diastereotopic α-CH2 in the complex 1, indicating that the rhodium carbon bond was formed regioselectively at the terminal primary CH2 unit (Fig. 1). No intermolecular addition product was formed as the β-hydroxyl proton observed for the reaction of allylbenzene was not found in the spectrum of the complex 1 (Scheme 2B).13 The independently synthesized rhodium alkyl complex by the reaction of (TSPP)RhI with 2-(iodomethyl)-2,3-dihydrobenzofuran resulted in the same 1H NMR spectrum as that of the complex 1. ESI-MS results (C53H33N4O13RhS4, m/z = 291.99903, calcd 291.99900; Fig. 1) also confirmed the structure of 1. The intramolecular –OH attack reaction of (TSPP)RhIII with 2-allylphenol to form 1 occurred through coordination of an alkene towards the rhodium center, followed by intramolecular nucleophilic attack of the phenoxyl group.11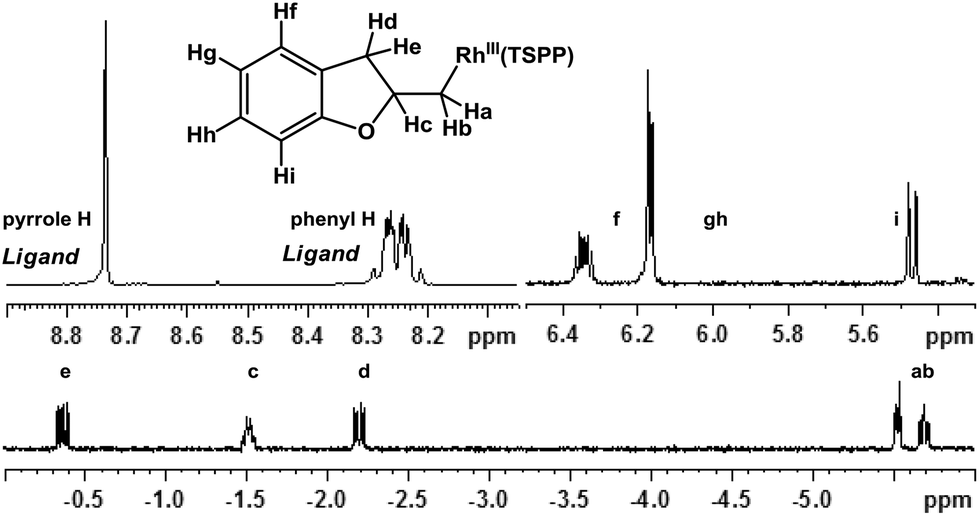 | ||
| Fig. 1 1H NMR spectrum of the complex 1 in CD3OD. | ||
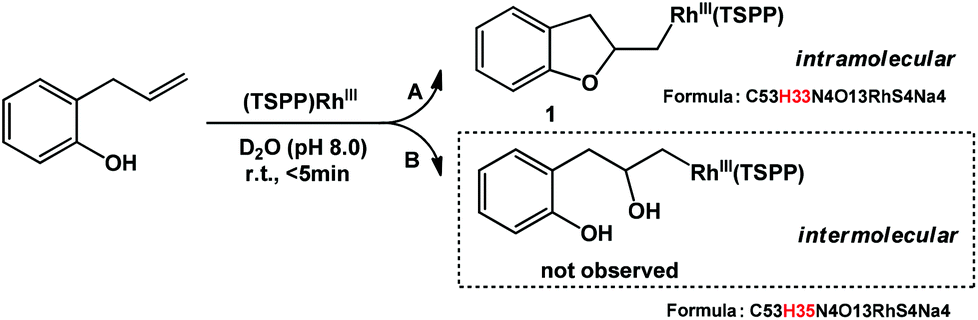 | ||
| Scheme 2 Reaction of (TSPP)RhIII with 2-allylphenol to form the complex 1. | ||
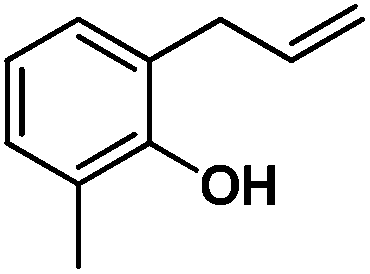
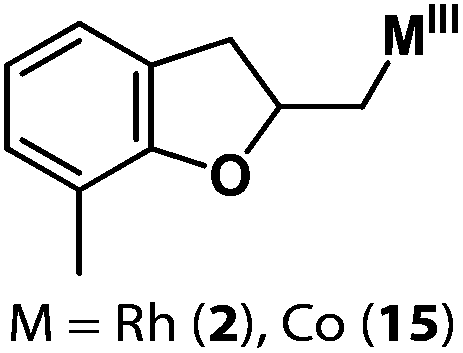
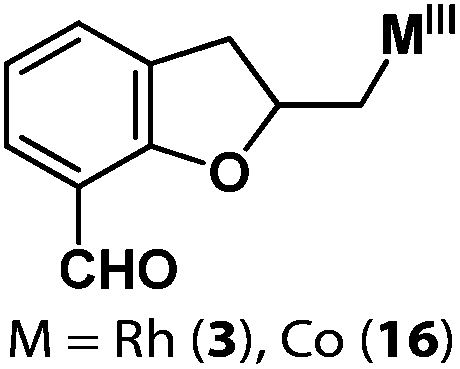
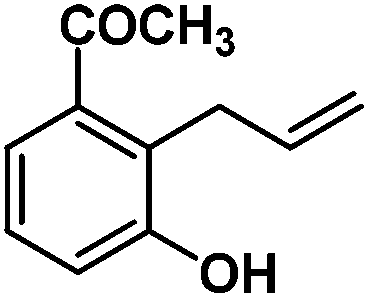
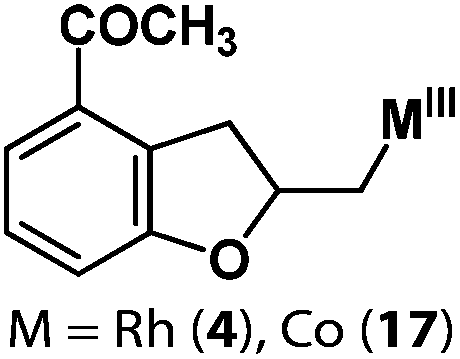
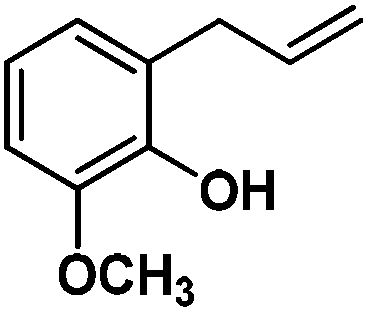
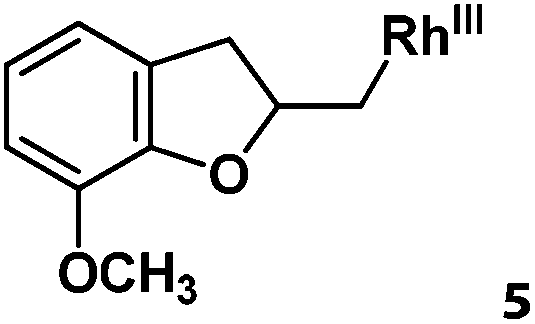
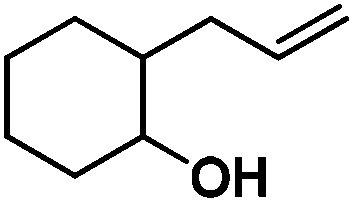
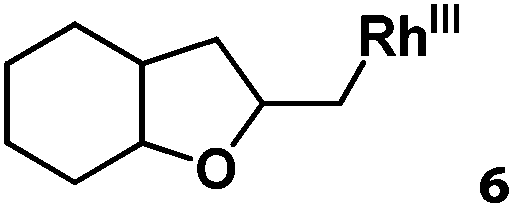
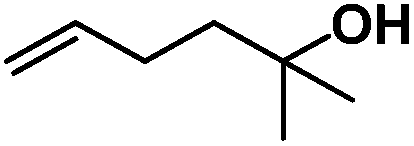
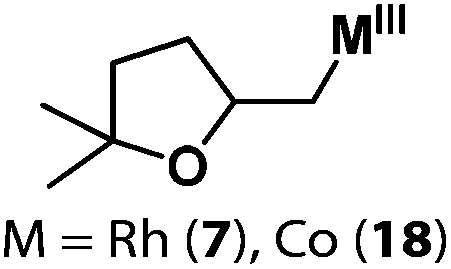
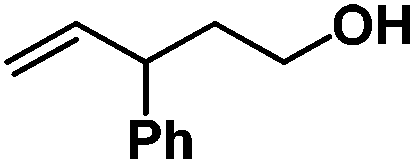
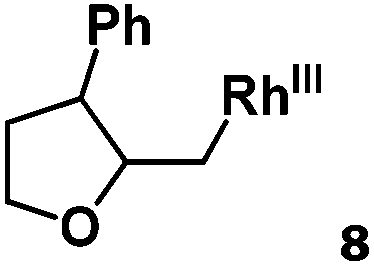
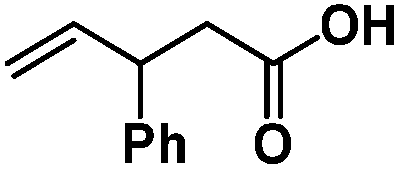
Following the same reactivity pattern, a series of phenyl substituted 2-allylphenol substrates (Table 1, entries 1–5) and a variety of alkenes with different nucleophiles (Table 1, entries 6–12) were observed to react rapidly and quantitatively with (TSPP)RhIII in water and methanol. Changing from the benzene ring to the cyclohexane ring, 2-allylcyclohexanol reacted with (TSPP)RhIII rapidly to form the cyclization product 6, indicating that the less nucleophilic alcoholic hydroxyl group could also perform intramolecular oxidative alkoxylation reaction (Table 1, entry 6). 2-Methylhex-5-en-2-ol also experienced a 5-exo-trig ring closure reaction with (TSPP)RhIII in water with formation of the complex 7 (Table 1, entry 7). The primary alcoholic hydroxyl group could also act as the nucleophile to form the intramolecular cyclization product 8 (Table 1, entry 8). A γ-lactone containing the rhodium alkyl complex 9 was obtained from a reaction of the 3-phenyl-4-pentenoic acid substrate with a carboxylic hydroxyl nucleophile (Table 1, entry 9). It is worth noting that 2-allylaniline also reacted with (TSPP)RhIII in an intramolecular way with the anilino group as the nucleophile (Table 1, entry 10).
| Entry | Substrate | Product | Yieldc |
|---|---|---|---|
| a Reaction conditions: (TSPP)RhIII (1.0 mmol L−1), alkene (10 equiv.), D2O (300 μL, pH 8.0 borate buffer), room temperature, <5 min. b Reaction conditions: (TSPP)CoIII (1.0 mmol L−1), alkene (10 equiv.), D2O (300 μL, pH 9.0 borate buffer), room temperature, <5 min. c Determined by 1H NMR. d 0.1 mL CD3OD was added to increase the solubility of the substrate. e Reactions were performed in CD3OD. | |||
| 1 |
|
|
1: >95% |
| 14: >95% | |||
| 2 |
|
|
2: >95% |
| 15: 73% | |||
| 3 |
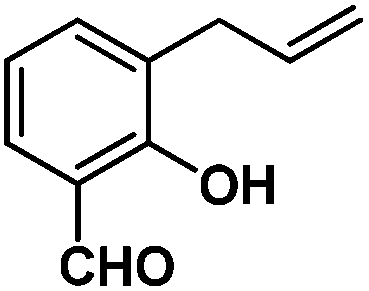
|
|
3: >95% |
| 16: 82% | |||
| 4 |
|
|
4: >95% |
| 17: 88% | |||
| 5 |
|
|
>95% |
| 6 |
|
|
>95% |
| 7 |
|
|
7: >95% |
| 18: >95% | |||
| 8 |
|
|
>95% |
| 9 |
|
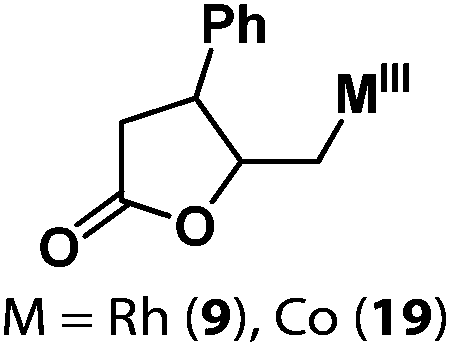
|
9: 86% |
| 19: 57% | |||
| 10 |
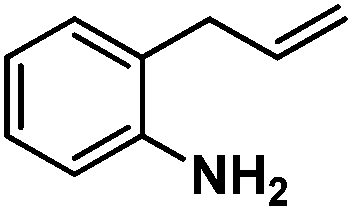
|
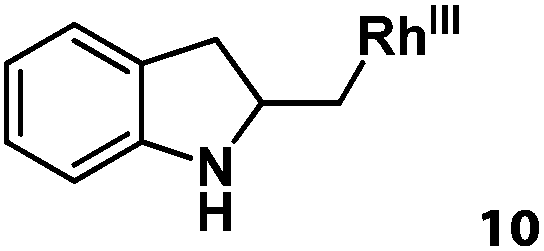
|
79% |
| 11 |
|
|
>95% |
| 12 |

|
|
>95% |
| 13 |
|
|
>95% |
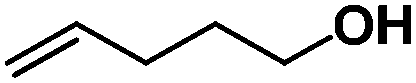
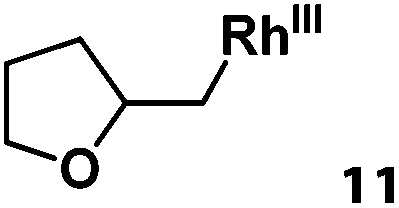
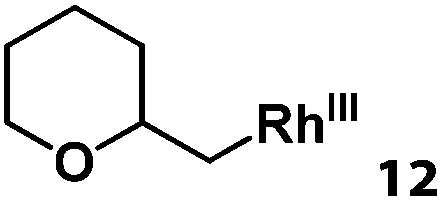
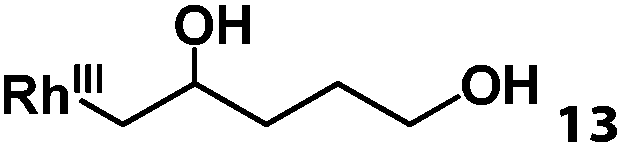
Previously, we reported the reactivity of pent-4-en-1-ol in water with rhodium porphyrins, and the reaction generated an exclusively intermolecular nucleophilic attack product 13.11 Comparison of entries 7 and 8 where the substrates share similar skeleton to pent-4-en-1-ol revealed the “pre-organizing” function of both phenyl and di-methyl groups which resulted in immediate intramolecular nucleophilic cyclization reaction. Although no cyclization was observed for pent-4-en-1-ol in water, reaction with (TSPP)RhIII in the methanol solvent rapidly produced the intramolecular alkoxylation rhodium alkyl complex 11 without observation of the 6-endo-trig product (Table 1, entry 11). Similar reactivity was also observed for hex-5-en-1-ol which underwent 6-exo-trig cyclization to form 12 with high regioselectivity (Table 1, entry 12). The difference between the intermolecular reactivity in water lies in the nucleophilicity increase of the alcoholic hydroxyl groups in methanol where the solvation effect is lower, and that the hydrophobic porphyrin ligand was away from the hydroxyl group on the alkyl chain.
The produced metal alkyl complexes of Table 1 were generally proposed as the key intermediates in Wacker-type oxidation and hydrofunctionalization of alkenes, although only a few studies reported direct observation of the intermediate.14 Formation of a new C–O bond in these reactions was thought to occur exclusively by attack of an oxygen nucleophile onto a metal-coordinated olefin.1–3 Recently, migratory insertion of a CC bond into an M–O bond was proposed as an alternative pathway.15 However, the four membered transition state of migration insertion would require unsaturation of the metal center with a vacant cis coordination site, which was not conveniently accessible for alkyl rhodium porphyrin complexes.11,16 Combined with previous studies on intermolecular (TSPP)RhIII mediated aerobic oxidation of alkenes, formation of alkyl rhodium species in Table 1 was proposed to occur via the intramolecular nucleophilic attack towards coordinated alkenes.
Production of unsaturated heterocyclic compounds
In the absence of air, the complex 1 was spontaneously transformed to (TSPP)RhI and 2-methylbenzofuran over 30 min at 333 K in water (Scheme 3). The resulting product was extracted with CDCl3 and identified by GC-MS and 1H NMR. The product was proposed to be formed by β-hydrogen elimination of the complex 1. The stoichiometric reactivity of a series of phenyl substituted 2-allylphenol substrates was examined (Scheme 3). Reactions with (TSPP)RhIII in pH 8.0 buffer solution were rapid and quantitative. The formed β-phenoxyalkyl rhodium porphyrins 2–5 underwent β-hydrogen elimination at 333 K under a nitrogen atmosphere to produce 2-methylbenzofurans quantitatively within 1 hour. The reaction exhibited good tolerance for a decent range of substituents including alkyl, alkoxyl, acyl, and formyl groups.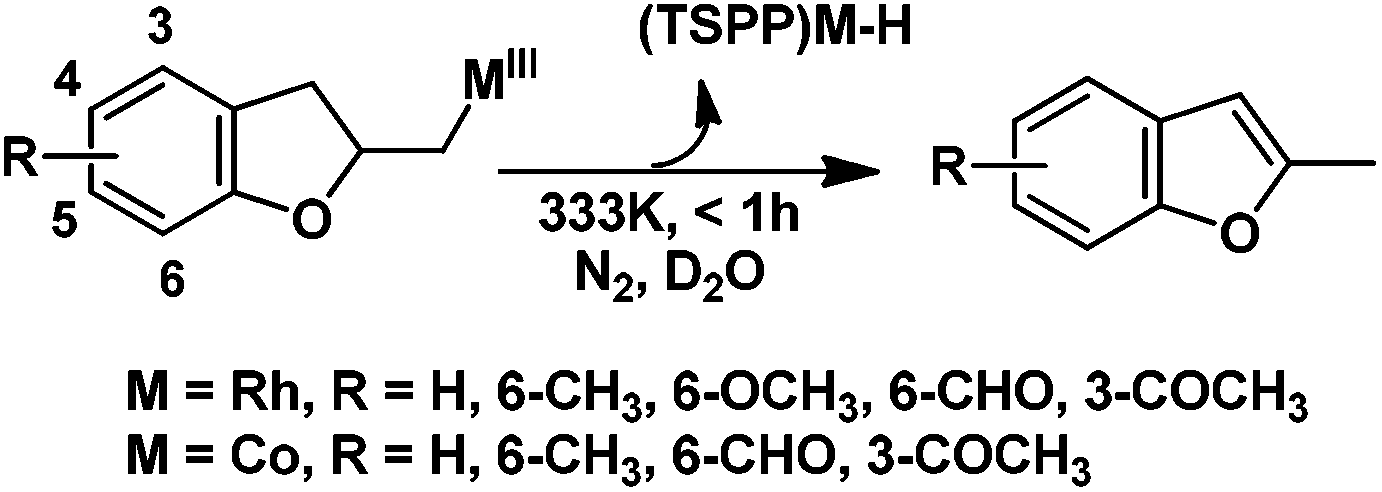 | ||
| Scheme 3 Formation of heterocyclic unsaturated compounds. See Table S1 in ESI† for detailed experimental conditions and results. | ||
(TSPP)CoIII mediated oxidative alkoxylation of alkenes
(TSPP)RhIII showed an unusual pathway for oxidative alkoxylation/cyclization of alkenes in water. The cheap metal complex (TSPP)CoIII was a much more attractive catalyst if this complex could react in the same reactivity pattern as that of (TSPP)RhIII. However, (TSPP)CoIII did not react with alkenes through the intermolecular pathway to form β-hydroxyl alkyl cobalt porphyrins. Fortunately, the intramolecular reaction occurred rapidly. Addition of 2-allylphenol to a pH 8.0 buffer solution of (TSPP)CoIII immediately led to formation of the cyclization product β-phenoxyalkyl cobalt complex 14 (Table 1, entry 1). Methyl, formyl, and acyl substituted allylphenols also showed similar reactivities (Table 1, entries 2–4). Rapid formation of alkyl cobalt species 18 and 19 was also observed by a reaction of 2-methylhex-5-en-2-ol and 3-phenyl-4-pentenoic with (TSPP)CoIII (Table 1, entries 7 and 9). These reactions provide rare examples of Co(III) mediated oxidation of alkenes,17 since both the interaction of Co(III) with alkenes and the Co(III)–C bonding are weak, which explained the unfavorable intermolecular formation of CoIII–CR2–CR2OH. The efficient formation of complexes 14–19 through the intramolecular nucleophilic addition pathway was ascribed to the entropy-driven effect. Upon heating to 333 K, complexes 14–17 gave an oxidative cyclization product 2-methylbenzofuran as observed for 1 (Scheme 3).The (TSPP)Rh-H species formed in elimination reactions in Scheme 3 occurs in a rapid dissociation equilibrium with (TSPP)RhI and H+. Rapid air oxidation of (TSPP)RhI to (TSPP)RhIII completes the Wacker-type oxidation cycle which provides the potential for aerobic catalytic intramolecular cyclization of olefins with exclusive regioselectivity (eqn (1)).18
| (TSPP)RhI + 1/2O2 + H2O ⇌ (TSPP)RhIII + 2OH− | (1) |
Conclusions
In summary, we have described an unusual pathway for intramolecular oxidative cyclization of substituted 2-allylphenols by (TSPP)RhIII and (TSPP)CoIII to form 2-methylbenzofuran derivatives in water. Key intermediates β-phenoxyalkyl rhodium/cobalt porphyrin complexes were observed and characterized. A series of substrates including unsaturated alcohols, carboxylic acid, and aniline also reacted with (TSPP)RhIII to form thermally stable β-hetero-functionalized alkyl rhodium porphyrins with exclusive regioselectivity. These complexes provide valuable insights into mechanistic studies of transition metal catalyzed Wacker-type oxidation of alkenes and a novel construction protocol for heterocycle synthesis. Developing new methods for further cleavage of the Rh–C bond and Co–C bond and catalytic intramolecular oxidative cyclization of alkenes is ongoing.Acknowledgements
This work was supported by the National Science Foundation of China (21171012).Notes and references
- (a) J. Smidt, W. Hafner, R. Jira, R. Sieber, J. Sedlmeier and A. Sabel, Angew. Chem., Int. Ed. Engl., 1962, 1, 80 CrossRef; (b) J. M. Takacs and X. T. Jiang, Curr. Org. Chem., 2003, 7, 369 CrossRef CAS; (c) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400 CrossRef CAS PubMed; (d) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS; (b) M. Beller and J. Seayad, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed; (c) E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563 CrossRef CAS PubMed; (d) R. Zhang, W. Y. Yu, H. Z. Sun, W. S. Liu and C.-M. Che, Chem. – Eur. J., 2002, 8, 2495 CrossRef CAS PubMed.
- (a) A. R. Chianese, J. Lee and M. R. Gagné, Angew. Chem., Int. Ed., 2007, 46, 4042 CrossRef CAS PubMed; (b) B. de Bruin, P. H. M. Budzelaar and A. W. Gal, Angew. Chem., Int. Ed., 2004, 43, 4142 CrossRef CAS PubMed; (c) R. Pryadun, D. Suumaran, R. Bogadi and J. D. Atwood, J. Am. Chem. Soc., 2004, 126, 12414 CrossRef CAS PubMed.
- (a) K. Utimoto, Pure Appl. Chem., 1983, 11, 1845 Search PubMed; (b) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS PubMed; (c) J. P. Wolfe, Synlett, 2008, 19, 2913 CrossRef.
- (a) F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104 RSC; (b) T. E. Mueller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef CAS PubMed; (c) K. D. Hesp and M. Stradiotto, ChemCatChem, 2010, 2, 1192 CrossRef CAS; (d) B. V. Popp and S. S. Stahl, Top. Organomet. Chem., 2007, 22, 149 CrossRef CAS; (e) L. Hintermann, Top. Organomet. Chem., 2010, 22, 123 CrossRef.
- (a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef; (b) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed.
- (a) H. Qian, X. Q. Han and R. A. Widenhoefer, J. Am. Chem. Soc., 2004, 126, 9536 CrossRef CAS PubMed; (b) X. W. Li, A. R. Chianese, T. Vogel and R. H. Crabtree, Org. Lett., 2005, 7, 5437 CrossRef CAS PubMed; (c) L. Coulombel, M. Rajzmann, J. M. Pons, S. Olivero and E. Duñach, Chem. – Eur. J., 2006, 12, 6356 CrossRef CAS PubMed; (d) Z. Zhang and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2007, 46, 283 CrossRef CAS PubMed; (e) C. Liu, C. F. Bender, X. Han and R. A. Widenhoefer, Chem. Commun., 2007, 3607 RSC; (f) S. Y. Seo, X. H. Yu and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 263 CrossRef CAS PubMed; (g) J. D. Neukom, N. S. Perch and J. P. Wolfe, Organometallics, 2011, 30, 1269 CrossRef CAS.
- (a) T. Hokosawa, S. Yamashita, S.-I. Murahashi and A. Sonoda, Bull. Chem. Soc. Jpn., 1976, 49, 3662 CrossRef; (b) T. Hosokawa, S. Miyaga, S.-I. Murahashi and A. Sonoda, J. Org. Chem., 1978, 43, 2752 CrossRef CAS; (c) Y. Uozumi, K. Kat and T. Hayashi, J. Am. Chem. Soc., 1997, 119, 5063 CrossRef CAS; (d) K. Muñiz, Adv. Synth. Catal., 2004, 346, 1425 CrossRef; (e) T. Hayashi, K. Yamasaki, M. Mimura and Y. Uozumi, J. Am. Chem. Soc., 2004, 126, 3036 CrossRef CAS PubMed; (f) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed.
- (a) B. R. James and D. V. Stynes, J. Chem. Soc., Chem. Commun., 1972, 1261 RSC; (b) B. B. Wayland, B. A. Woods and R. Pierce, J. Am. Chem. Soc., 1982, 104, 302 CrossRef CAS; (c) Y. Aoyama, T. Yoshida and H. Ogoshi, Organometallics, 1986, 5, 168 CrossRef CAS; (d) J. E. Anderson, C. L. Yao and K. M. Kadish, J. Am. Chem. Soc., 1991, 113, 5305 CrossRef; (e) J. P. Collman and R. Boulatov, Inorg. Chem., 2001, 40, 560 CrossRef CAS PubMed; (f) X. Fu and B. B. Wayland, J. Am. Chem. Soc., 2005, 127, 16460 CrossRef CAS PubMed; (g) X. Fu, S. Li and B. B. Wayland, J. Am. Chem. Soc., 2006, 549 Search PubMed; (h) K. S. Chan, X. Z. Li, W. I. Dzik and B. de Bruin, J. Am. Chem. Soc., 2008, 130, 2051 CrossRef CAS PubMed; (i) C.-M. Che and J.-S. Huang, Chem. Commun., 2009, 3996 RSC.
- M. S. Sanford and J. T. Groves, Angew. Chem., Int. Ed., 2004, 43, 588 CrossRef CAS PubMed.
- (a) J. Zhang, S. Li, X. Fu and B. B. Wayland, Dalton Trans., 2009, 3661 RSC; (b) J. Zhang, B. B. Wayland, L. Yun, S. Li and X. Fu, Dalton Trans., 2010, 39, 477 RSC; (c) B. Wu, J. Zhang, L. Yun and X. Fu, Dalton Trans., 2011, 40, 2213 RSC.
- (a) K. Hori, H. Kitagawa, A. Miyoshi, T. Ohta and I. Furukawa, Chem. Lett., 1998, 11, 1083 CrossRef; (b) T. Mitsudome, T. Umetani, N. Nosaka, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 481 CrossRef CAS PubMed.
- Reaction of (TSPP)RhIII and allylbenzene was performed in H2O. The formed product ((TSPP)Rh–CH2CH(OH)CH2C6H5) was purified and dissolved in anhydrous DMSO-d6 to observe the β-hydroxyl resonance (0.04 ppm, d, 3JH–H = 5.6 Hz). Similar workup was used for 2-allylphenol and no hydroxyl signal was observed in the 1H NMR spectrum of 1 in DMSO-d6.
- (a) K. A. Woerpel and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 1888 CrossRef; (b) C. Hahn, Chem. – Eur. J. 2004, 10, 5888 Search PubMed.
- P. S. Hanley and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 8510 CrossRef CAS PubMed.
- (a) J. P. Collman, A. S. Chien, T. A. Eberspacher and J. I. Brauman, J. Am. Chem. Soc., 1998, 120, 425 CrossRef CAS; (b) I. H. Grate and G. N. Schrauzer, J. Am. Chem. Soc., 1979, 101, 4601 CrossRef; (c) G. N. Schrauzer and I. H. Grate, J. Am. Chem. Soc., 1981, 103, 541 CrossRef CAS; (d) K. W. Mak and K. S. Chan, J. Am. Chem. Soc., 1998, 120, 9686 CrossRef CAS; (e) K. W. Mak, F. Xue, T. C. W. Mak and K. S. Chan, J. Chem. Soc., Dalton Trans., 1999, 3333 RSC; (f) S. K. Yeung and K. S. Chan, Organometallics, 2005, 24, 2561 CrossRef CAS.
- (a) H. Sugimoto, M. Nagano, Z. Yoshida and H. Ogoshi, Chem. Lett., 1980, 9, 521 CrossRef; (b) M. T. Reetz and K. Töllner, Tetrahedron Lett., 1995, 36, 9461 CrossRef CAS; (c) T. Hayashi, K. Okazaki, N. Urakawa, H. Shimakoshi, J. L. Sessler, E. Vogel and Y. Hisaeda, Organometallics, 2001, 20, 3074 CrossRef CAS.
- X. Fu and B. B. Wayland, J. Am. Chem. Soc., 2004, 126, 2623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qi00066h |
| This journal is © the Partner Organisations 2014 |