
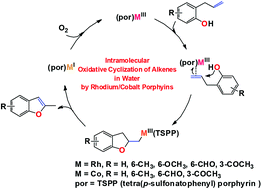
Intramolecular oxidative cyclization of alkenes by rhodium/cobalt porphyrins in water†
Abstract
Intramolecular oxidative cyclization of alkenes provides a unique pathway to obtain unsaturated heterocyclic compounds. The rhodium(III)/cobalt(III) tetra(p-sulfonatophenyl)porphyrin ((TSPP)MIII) in water was observed to mediate the intramolecular oxidative functionalization of alkenes to a series of unsaturated oxygen heterocyclic compounds.
- This article is part of the themed collection: Celebrating 110th Anniversary of Chemistry at Peking University
 Please wait while we load your content...
Please wait while we load your content...

