 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards chromate-free corrosion inhibitors: structure–property models for organic alternatives†
D. A.
Winkler
*ad,
M.
Breedon
bc,
A. E.
Hughes
c,
F. R.
Burden
ad,
A. S.
Barnard
b,
T. G.
Harvey
c and
I.
Cole
*c
aCSIRO Materials Science & Engineering, Clayton, Australia. E-mail: dave.winkler@csiro.au
bVirtual Nanoscience Laboratory, CSIRO Materials Science & Engineering, Parkville, Australia
cEnvironmental Surface Design, CSIRO Materials Science & Engineering, Clayton, Australia. E-mail: ivan.cole@csiro.au
dMonash Institute of Pharmaceutical Sciences, Parkville, Australia
First published on 6th May 2014
Abstract
Progressive restrictions on the use of toxic chromate-based corrosion inhibitors present serious technical challenges. The most critical of these is the lack of non-toxic ‘green’ alternatives that offer comparable performance, particularly on corrosion-prone aluminium alloys such as the 2000 and 7000 series. In this study we used computational modelling methods to investigate the properties of a range of small organic, potentially safer inhibitors and their interactions with technologically relevant alloy surfaces. We have generated robust and predictive computational models of corrosion inhibition for a structurally related data set of organic compounds from the literature. Our studies have correlated molecular features of the inhibitor molecules with inhibition and identified those features that have the greatest impact on experimentally determined corrosion inhibition. This information can be used to drive guided decision making for in silico or experimental screening of molecules for their corrosion inhibition efficiency, while considering more carefully their environmental consequences.
1. Introduction
Traditionally, many metallic structures in corrosive environments have been protected by the use of chromates, as corrosion inhibitors in paint films, or as conversion coatings on the surface of metals or alloys.1 Chromates are very effective inhibitors but recent studies have shown that they can be occupational ‘hot spot’ pollutants posing significant risk to workers involved in their production, or as by-products from operations such as paint removal and metal preparation. Recent epidemiology data from a large study of chrome chemical production workers found the excess lifetime risk of dying from lung cancer from occupational exposures to be 255 per 10002 or 255![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per million, massively larger than the acceptable risk of 1 death per million. Consequently, chromates are considered to present an unacceptable health risk3 and are progressively being limited or withdrawn from service by national legislation. Considerable effort has been expended in looking for non toxic alternatives to chromates.4 Increasingly,5–8 experimental approaches are being combined with molecular modelling in an effort to find new, more benign inhibitors. Modelling studies have predominantly utilised quantum chemical methods based on density functional theory (DFT) together with statistical or machine learning modelling techniques embodied in the quantitative structure–property relationships (QSPR) method.9 There is a recent history of successful application of both electronic structure simulations10,11 and QSPR9,12–17 to the prediction of toxicity in functional materials and coatings.
000 per million, massively larger than the acceptable risk of 1 death per million. Consequently, chromates are considered to present an unacceptable health risk3 and are progressively being limited or withdrawn from service by national legislation. Considerable effort has been expended in looking for non toxic alternatives to chromates.4 Increasingly,5–8 experimental approaches are being combined with molecular modelling in an effort to find new, more benign inhibitors. Modelling studies have predominantly utilised quantum chemical methods based on density functional theory (DFT) together with statistical or machine learning modelling techniques embodied in the quantitative structure–property relationships (QSPR) method.9 There is a recent history of successful application of both electronic structure simulations10,11 and QSPR9,12–17 to the prediction of toxicity in functional materials and coatings.
DFT methods can derive a range of molecular properties such as HOMO and LUMO energies, fundamental gap ΔE, chemical potential μ, electronegativity χ, and chemical hardness η, generally in vacuo. A number of published reports have claimed to identify trends or statistical correlations between these types of electronic properties and experimentally measured corrosion inhibition values. However, a 2008 review by Gece18 concerning the application of DFT methods concluded that, “calculations performed with inaccurate methods or with an insufficient dataset can easily lead to erroneous results”. Indeed many of the published studies have been undertaken on very small data sets and without adequate consideration of the presence of solvent, ions, or other aspects of the complex chemical environment in which corrosion and inhibition occur.
One of the most interesting classes of inhibitors is the substituted heterocyclic class of organic compounds. We undertook the research outlined in this paper to determine whether or not correlations between quantum chemical properties and corrosion inhibition were valid for this promising class of ‘green’ corrosion inhibitors, or whether they indeed exist. We also investigated alternative ways of modelling the relationships between the molecular properties of small organic corrosion inhibitors and performance under ‘real world’ experimental conditions. Relative to other published corrosion inhibition modelling studies this work makes two significant contributions to the search for more benign, small molecule replacements for toxic chromates:
• it uses a well-designed and relatively large (for a corrosion study) experimental data set. We used the experimental data published by Harvey et al.19 comprising 28 organic inhibitors, many based on substituted heterocyclic structures of different types (‘the Harvey data set’). The inhibition efficiencies within the Harvey data set varied from −175% (enhanced corrosion) to 98% (almost complete inhibition of corrosion) and were measured from mass loss data over 28 day immersion tests in saline solution at an inhibitor concentration of 1 mM.
• it uses an extensive pool of molecular properties related to atom types, functional groups, and molecular connectivity calculated from structural features of the inhibitor molecules. Molecular level properties are commonly called molecular descriptors.
We chose these data to minimize false or chance correlations due to limited experimental data and limited range of inhibition measurements. The sparse feature selection methods we adopted also minimizes the likelihood of chance correlations due to choosing subsets of parameters incorrectly from larger pools of possibilities. An important overall aim of these experiments was to establish whether predictive models of corrosion inhibition could be generated that could help accelerate the search for safe and effective alternatives to chromates.
2. Materials and methods
2.1 Corrosion data
The data on percent inhibition (or acceleration) of corrosion were taken from Harvey et al.19 and was based on mass loss over 28 days immersion in saline. The aluminium alloys used in this study were AA2024-T3 and AA7075-T6. The compositions were determined by ICP as AA2024-T3 (Cu 5.3%, Mg 1.6%, Mn 0.6%, Fe 0.2%, Zn <0.1%) and AA7075-T6 (Cu 1.4%, Mg 2.4%, Mn <0.1%, Fe 0.2%, Zn 5.4%).19 AA2024-T3 was much higher in copper than 7075, while 7075 was much higher in zinc. Iron and magnesium levels were comparable in both alloys.Additional immersion tests on AA1150 series Al (nearly pure Al) and pure Cu with some of the small molecule inhibitors used in our study found very little interaction with Al (which has a low affinity for S and N).20 Cu by contrast was affected by several of the organic compounds tested, forming coloured solutions and/or precipitates of corrosion products. Although the behaviour of an intermetallic may be different to that of bulk Cu, these experiments show that Cu is interacting strongly with some of the inhibitor molecules.
2.2 Speciation
The identities of the 28 small organic molecules from Harvey et al.19 are summarized in Table 1, and their chemical structures shown in Fig. 1. The speciation of the inhibitors was calculated using the SPARC method.21 SPARC uses relatively simple computational algorithms to estimate pKas of organic molecules from their structures. Structures are broken at each essential single bond into functional units that have intrinsic properties. Acidic and basic reaction centres are identified, and the impact of attached structural features on pKa is estimated using perturbation theory. Structures of the inhibitors were input as SMILES strings to the SPARC program, and the relative populations of ionized and neutral species were calculated at pH 7. Molecular and DFT-based descriptors were calculated for neutral and ionized states where relevant.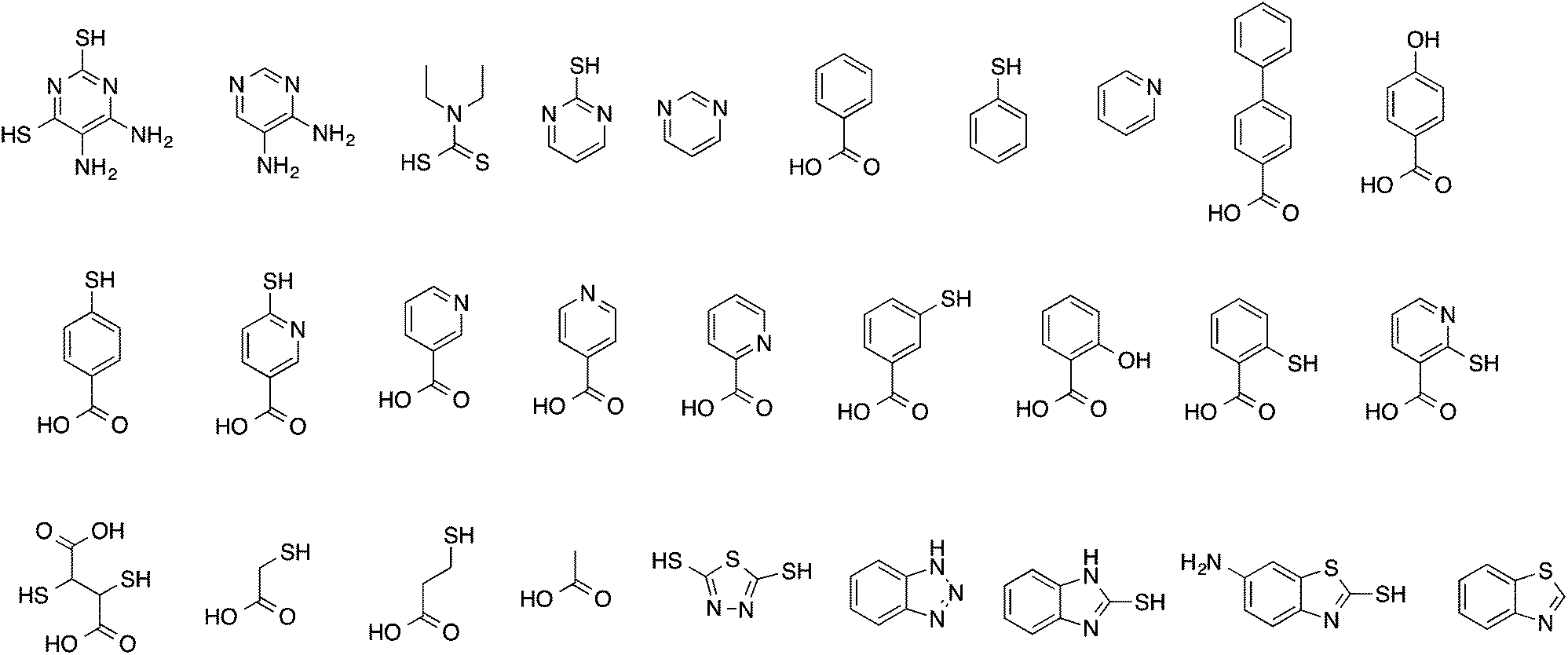 | ||
| Fig. 1 Structures of inhibitors in the data set. | ||
| Inhibitor | AA2024-T3 | AA7075-T6 | Speciesa |
|---|---|---|---|
| a S−, COO− denotes ionized thiol or carboxylate; N+ means protonated nitrogen atom; N+S− is a zwitterionic form. Sodium salts of anions were used where relevant. | |||
| 2-Mercaptobenzimidazole | 90 | 84 | N+S− |
| 2-Mercaptobenzothiazole | 95 | 91 | S− |
| 2-Mercaptopyrimidine | 89 | 50 | S−, N+S− |
| 2,5-Dimercapto-1,3,4-thiadiazole | 26 | −32 | 2 × S− |
| 4,5-Diamino-2,6-dimercaptopyrimidine | 87 | 80 | N+S−S− |
| 4,5-Diaminopyrimidine | 47 | 84 | Neutral |
| 6-Amino-2-mercaptobenzothiazole | 89 | 94 | S− |
| Benzotriazole | 98 | 92 | Neutral |
| 2,3-Mercaptosuccinate | 82 | 48 | 2 × COO− |
| 4-Phenylbenzoate | −72 | −143 | COO− |
| 6-Mercaptonicotinate | 94 | 86 | N+S−, COO− |
| 2-Mercaptobenzoate | 88 | 80 | COO− |
| 2-Mercaptonicotinate | 83 | 70 | N+S−, COO− |
| 3-Mercaptobenzoate | 16 | −22 | S−, COO− |
| 4-Hydroxybenzoate | −34 | −56 | COO− |
| 4-Mercaptobenzoate | 97 | 76 | S−, COO− |
| Acetate | −12 | 15 | COO− |
| Diethyldithiocarbamate | 97 | 96 | S− |
| Isonicotinate | −12 | −45 | COO− |
| Mercaptoacetate | 96 | 83 | COO− |
| Mercaptopropionate | 100 | 31 | COO− |
| Nicotinate | −107 | −91 | COO− |
| Picolinate | 58 | 14 | COO− |
| Salicylate | −175 | −89 | COO− |
| Pyridine | −139 | −150 | Neutral |
| Pyrimidine | −153 | −220 | Neutral |
| Benzoate | −80 | −62 | COO− |
| Thiophenol | 93 | 87 | S− |
The small organic molecules in the Harvey data set exhibit significant chemical diversity and a wide range of speciation behaviour, depending on the number and nature of their ionisable groups. In some cases the identity of the organic species was clear at the neutral pH. However, some of the heteroaromatic compounds, and indeed, some inhibitors that contained both COOH and SH acidic moieties, exhibited quite complex speciation. In some instances there were as many as 5–6 coexisting species at pH 7. Given that these may have different affinities for metal surfaces and clearly different molecular properties as calculated by DFT and other methods, it was useful to identify the main species that exist at the experimental pH. The dominant speciation, and the corrosion performance for the two aerospace alloys for the Harvey data set are also summarized in Table 1.
The speciation of some of the inhibitors was quite complex. Harvey et al.19 assumed that some molecules contained a single acidic moiety when they generated sodium salts by adding an appropriate number of moles of sodium carbonate. We have assumed that the effects of incomplete neutralization and salt formation were minimal.
2.3 DFT calculated molecular properties
The DFT derived molecular descriptors generated were:| Ionization potential (IP) = EN−1 − EN | (1) |
| Electron affinity (EA) = EN − EN+1 | (2) |
| Absolute hardness (η) = (IP − EA)/2 | (3) |
| Chemical potential (μ) = −(IP + EA)/2 | (4) |
| Mulliken electronegativity (χ) = (IP + EA)/2 | (5) |
These molecular identifiers were obtained for each of the 28 inhibitors calculated by DFT using the Spanish Initiative for Electronic Simulations with Thousands of Atoms (SIESTA)22 and Gaussian packages.23 The exchange correlation functional of Perdew–Burke–Ernzerhof (PBE)24 with a double zeta plus polarization (DZP) basis set and cut off energy of 500 Ry was employed for all SIESTA calculations. All norm-conserving pseudopotentials were used as supplied with the SIESTA code without further modification. Structures were converged in a 30 × 30 × 30 Å supercell, until the residual forces on atoms was less than 0.01 eV Å−1 and the total energy difference between SCF steps was less than 10−4 eV. For comparison, Gaussian09 calculations were performed as all electron calculations utilising the 6-311++G** basis set; also utilising the PBE exchange correlation functional.
2.4 Quantitative structure–inhibition relationship studies
The molecules in the Harvey data set were constructed using Sybyl ×2.0 (Certera Limited). The structures were energy minimized using the Tripos force field. They were used to calculate a range of molecular descriptors such as the log of the octanol–water partition coefficient (a measure of molecular lipid solubility), molecular surface area, volume, molar refractivity (size and polarizability), polar surface area, numbers of hydrogen bond donors and acceptors. The structures were also used to generate a large variety of computed molecular descriptors using the DRAGON program25 and our in-house modelling package, BioModeller.26–28 We selected relevant descriptors from a pool of 173 in-house, and 194 Dragon descriptors. We also generated functional group descriptors that describe or quantify chemical moieties or fragments in molecules. These were: the number of sulfur atoms, number of ionized sulfur atoms, number of ionized COOH groups, number of rings, number of heteroaromatic nitrogen groups, and total molecular charge. Descriptors were calculated for neutral and ionized states at pH 7 where relevant.Models relating molecular properties to corrosion inhibition were constructed using the BioModeller software. The Bayesian modelling methods embodied in the BioModeller package have been described in detail elsewhere.27,29–35 Both linear and nonlinear models were generated. Linear models used sparse linear regression methods. The nonlinear models used a Bayesian regularized neural network26,29,31,32,35,36 that automatically controls model complexity to optimize the predictive performance of the models. The neural network training was stopped when the Bayesian evidence for the models was maximal. Generally two or three hidden layer nodes were employed in a three layer feed forward neural network; these types of models are relatively insensitive to the neural network architecture. The input and output layers nodes contained linear transfer functions, and the hidden layer nodes (where the computation is carried out) employed sigmoidal transfer functions.
Although models derived from Bayesian regularized neural networks do not strictly require a test or validation set, the predictive power of the models was assessed,37,38 nonetheless, by partitioning the data set into a training set (80% of the compounds) and test set (20% of the compounds). The performance of the models was assessed using the standard error of prediction of the training and test sets. Other statistical measures of merit were also calculated although these are not as robust (more influenced by the size of the data set and number of descriptors) as the standard error.
3. Results and discussion
3.1 Relationship between corrosion inhibitors for the two aluminium alloys
The inhibition results over 28 days immersion in saline, for the two alloys AA2024-T3 and AA7075-T6, correlate strongly with each other (r2 = 0.84, Fig. 2). The corrosion inhibition exhibited by the 28 compounds was 10% lower on average for the AA7075-T6 alloy than for the AA2024-T3 alloy.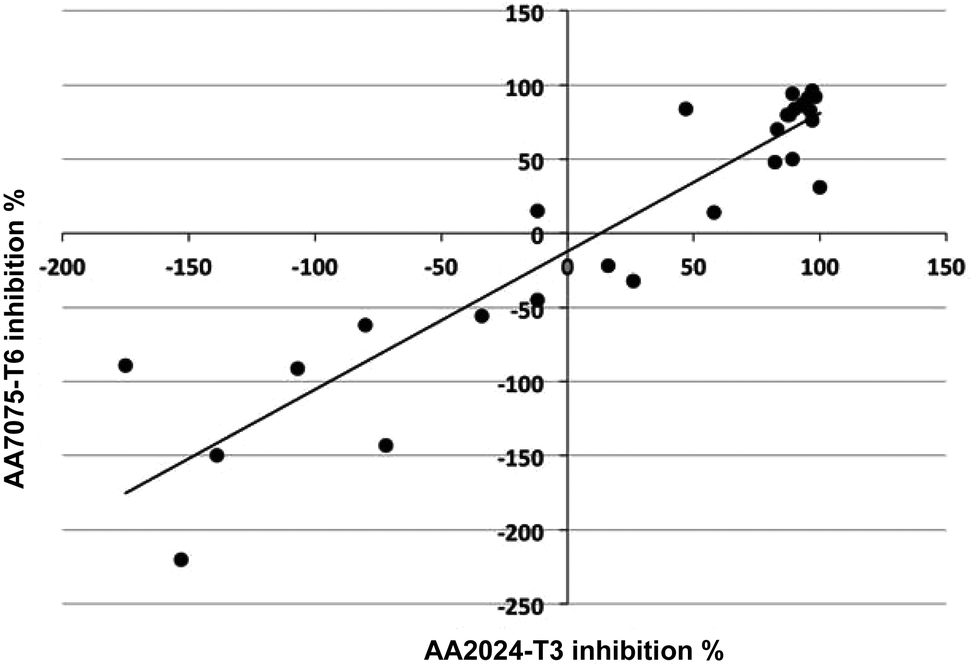 | ||
| Fig. 2 Correlation between corrosion inhibition on AA7075-T6 and on AA2024-T3 for 28 small organic inhibitors. | ||
Inspection of the inhibition results for the two alloys reveals that sulfur-containing ligands are almost uniformly more effective inhibitors of corrosion on the AA2024-T3 alloy than the AA7075-T6 alloy (e.g. 2-mercaptopyrimidine, 3-mercapto-benzoate, 2-mercaptonicotinate, 2,3-dimercapto-succinate, mercaptoacetate, mercaptopropionate, 2,5-dimercapto-1,3,4-thiadiazole). This may be due to the higher Cu content of this alloy compared to AA7075-T6. Conversely, compounds that do not contain sulfur are often more corrosive on the AA7075-T6 alloy than on the AA2024-T3 alloy (e.g. pyridine, pyrimidine, 4-phenoxybenzoate, 4-hydroxybenzoate), which may be reflective of the high Zn content found in AA7075-T6 alloy, compared with the AA2024-T3 alloy (richer in Cu).39
3.2 Relationship between corrosion inhibition and DFT properties
We calculated DFT-based and other molecular descriptors described below for two scenarios: assuming the molecules were neutral; assuming they were speciated at pH 7 according to Table 1. Initial modelling investigations aimed at determining the best sets of descriptors for generating robust and predictive inhibition models indicated, somewhat surprisingly, that speciation has relatively little effect on model quality. This is consistent with experimental corrosion testing carried out in CSIRO laboratories.40Other work has shown that, whether or not speciation was included, there was essentially no correlation between ionization potential, HOMO or LUMO energies, or any other quantum chemically-derived descriptors and corrosion efficiency.41 A significant number of literature reports5,42–50 claim that the frontier orbital energies and molecular properties derived from such energies are related to the corrosion inhibition. However, many of these studies used a very small number of inhibitors, in some cases as few as four, making the probability of chance correlations high. They also ignore the effects of solvent, ions and salts, speciation, and the presence of a metal surfaces, as the calculations would not be tractable if these were included. As discussed in section 3.4, molecular descriptors derived from the in vacuo DFT calculations on the Harvey data set were identified to be among the least relevant descriptors for generating predictive models of corrosion inhibition. The correlations between the DFT and molecular descriptors, and the corrosion inhibition for the two alloys are listed in ESI.†
3.3 Machine learning-based quantitative structure–inhibition modelling
We used the sparse feature selection capabilities of BioModeller to select the most relevant subset of descriptors from a large pool in a context dependent manner. As the data set was of moderate size we used all of the data in the feature selection process. The machine learning methods have been shown to generate robust models on small to moderate sized data sets without the need for a test set,29 although we chose to use a test set in this study.We generated statistically significant models that could predict the corrosion inhibition of compounds in an external test set using DRAGON descriptors and in-house chemically intuitive descriptors. We found a set of between 7 and 9 descriptors in each descriptor family could generate linear and nonlinear models that could make good, quantitative predictions of the degree of inhibition of molecules in the data set. As mentioned above, descriptors based on the speciated form of the inhibitors at pH 7 generated models of similar quality to those assuming neutral inhibitors, so the results for the neutral form of the molecules are reported here.
| Model | r 2train | SEE % | r 2test | SEP % | N desc | N eff |
|---|---|---|---|---|---|---|
| a Descriptors used were: nR09, nBnz, C-027, BEHm7, HOMT, C-044, O-057, S-106. | ||||||
| MLREM | 0.87 | 39 | 8 | 9 | ||
| MLREM | 0.86 | 43 | 0.91 | 36 | 8 | 9 |
| 20% test | ||||||
| BRANNGP | 0.83 | 24 | 8 | 11 | ||
| 2 nodes | ||||||
| BRANNGP | 0.77 | 24 | 0.88 | 32 | 8 | 11 |
| 2 nodes | ||||||
| 20% test | ||||||
| Model | r 2train | SEE % | r 2test | SEP % | N desc | N eff |
|---|---|---|---|---|---|---|
| a Descriptors used were: SH, S, BCGM2, BCGM4, BCGM5, A11, A31. | ||||||
| MLREM | 0.61 | 64 | 7 | 8 | ||
| MLREM | 0.60 | 71 | 0.79 | 31 | 7 | 8 |
| 15% test | ||||||
| BRANNGP | 0.79 | 31 | 7 | 11 | ||
| 2 nodes | ||||||
| BRANNGP | 0.74 | 35 | 0.99 | 23 | 7 | 10 |
| 2 nodes | ||||||
| 15% test | ||||||
The nonlinear models were sparse, using only 10–11 effective weights in the model and employing 7–8 descriptors, and gave superior prediction to the linear models. The standard error of prediction for the test set was 23% for the nonlinear model compared to 31% for the linear model using atomistic and functional group descriptors.
The Dragon descriptors also generated predictive models of corrosion inhibition. The linear and nonlinear models could predict the inhibition of compounds in the training set with a standard error of 43% and 24% and 36% and 32% for the test set. The quality of the prediction of the training and test set for the best models employing Dragon and in-house descriptors is illustrated in Fig. 3 and 4.
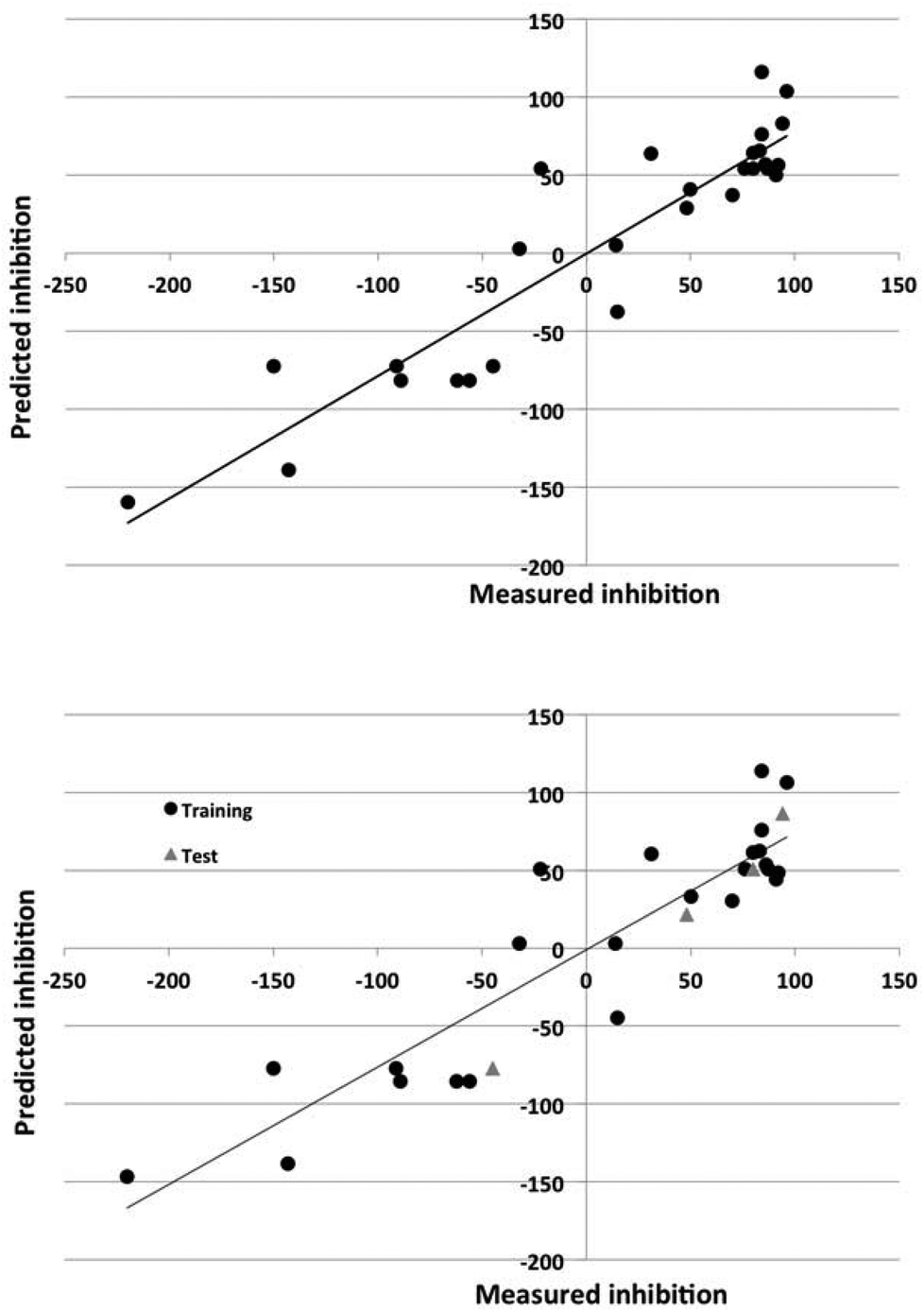 | ||
| Fig. 3 Observed versus predicted corrosion inhibition for 7075 alloy for the nonlinear model using DRAGON descriptors. Top panel: model for entire data set. Bottom panel: data set split into training (circles) and test (triangles) sets. | ||
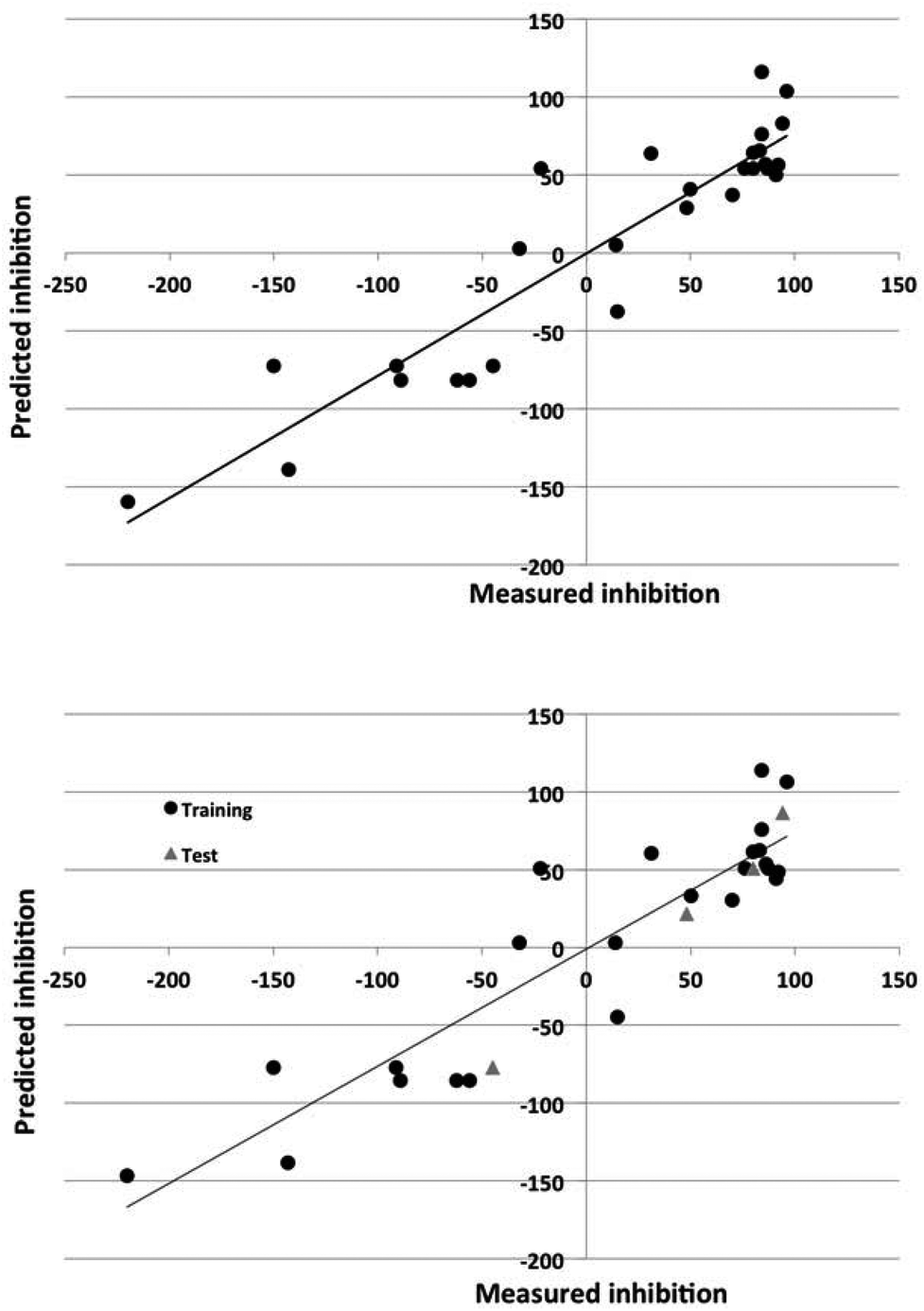 | ||
| Fig. 4 Observed versus predicted corrosion inhibition for 7075 alloy for the nonlinear models using the in-house intuitive descriptors. Top panel: model for entire data set. Bottom panel: data set split into training (circles) and test (triangles) sets. | ||
The nonlinear models could account for 70–90% of the variance in the data. The ability of the models to predict the degree of inhibition of the external test set compounds is good, as Fig. 3 and 4 also show.
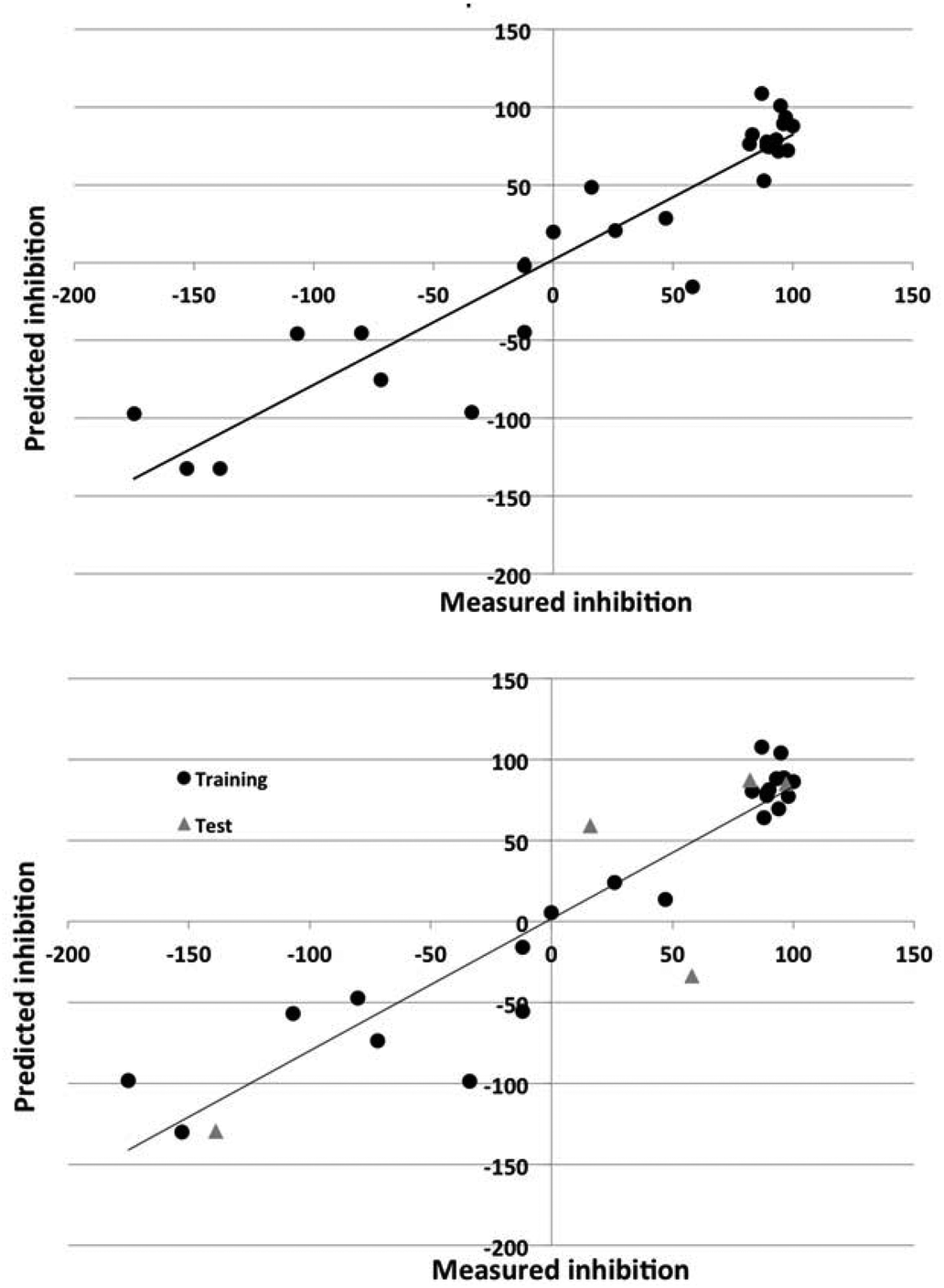 | ||
| Fig. 5 The observed versus predicted corrosion inhibition for 2024 alloy for the nonlinear model using DRAGON descriptors. Top panel: model for entire data set. Bottom panel: data set split into training (circles) and test (triangles) sets. | ||
 | ||
| Fig. 6 The observed versus predicted corrosion inhibition for 2024 alloy for the nonlinear models using the in-house intuitive descriptors. Top panel: model for entire data set. Bottom panel: data set split into training (circles) and test (triangles) sets. | ||
| Model | r 2train | SEP % | r 2test | SEP % | N desc | N eff |
|---|---|---|---|---|---|---|
| a Descriptors used were: nR09, nBnz, C-027, BEHm7, HOMT, C-044, O-057, S-106. | ||||||
| MLREM | 0.82 | 45 | 8 | 9 | ||
| MLREM | 0.83 | 46 | 0.80 | 49 | 8 | 9 |
| 20% test | ||||||
| BRANNGP | 0.80 | 30 | 8 | 11 | ||
| 2 nodes | ||||||
| BRANNGP | 0.81 | 28 | 0.74 | 45 | 8 | 11 |
| 2 nodes | ||||||
| 20% test | ||||||
| Model | r 2train | SEE % | r 2test | SEP % | N desc | N eff |
|---|---|---|---|---|---|---|
| a Descriptors used were: SH, S, BCGM2, BCGM4, BCGM5, A11, A31. | ||||||
| MLREM | 0.62 | 64 | 7 | 8 | ||
| MLREM | 0.67 | 65 | 0.42 | 94 | 7 | 8 |
| 20% test | ||||||
| BRANN | 0.75 | 36 | 7 | 9 | ||
| 2 nodes | ||||||
| BRANN | 0.76 | 36 | 0.69 | 46 | 7 | 9 |
| 2 nodes | ||||||
| 20% test | ||||||
As Tables 4 and 5 show, the DRAGON descriptors generated models with higher statistical significance than did the atomistic and functional group descriptors for the linear models. The nonlinear models were of similar predictive power.
The nonlinear models were sparse, using only 7–8 molecular descriptors and 9–11 effective weights in the models, and gave superior prediction to the linear models (SEP values of 45% versus 49% (Dragon descriptors) and 46% versus 94% (in-house intuitive descriptors).
It is clear from Fig. 5 that the DRAGON descriptors generated models that represented the data more evenly across the inhibition range. The atomistic and functional group descriptors tended to classify compounds either as inhibitors or non-inhibitors/accelerants as shown by the clustering in the right hand side of Fig. 6. As discussed previously, this is exacerbated by the rather uneven distribution of inhibition values across the range compared to those for AA7075-T6.
3.4 Interpretation of the models
The small organic molecules that accelerate corrosion rather than inhibit it tend to be fairly strong organic acids (low pKa values for either the COOH or SH moieties, see Table 1). This may provide a partial explanation for the deleterious effects of some of these molecules, particularly for the zinc-rich AA7075-T6 alloy. These types of molecules may also destabilize the oxide layer on the surface of the metal, or generate metal carboxylates, thus accelerating corrosion. The corrosive effect of organic compounds is quite complex, and has been reviewed by Heitz.51 He illustrates the potential for protic organic compounds in particular to accelerate rather than inhibit corrosion.The quantitative structure–inhibition relationships models show that a relatively small number of molecular properties affected the inhibition. Some of these descriptors are arcane and hard to interpret. It appears that sulfur atoms can in some cases ameliorate corrosive potential. It is clear by inspection that in many cases the presence of a sulfur atom, particularly as an ionized –SH moiety combined with proximity to a heteroatom in a ring, generates compounds with very good corrosion inhibition performance. The relevant descriptors for models using DRAGON descriptors were: the number of rings containing 9 heavy atoms (e.g. benzimidazole) (nR09); the number of benzene-like rings (nBnz); the number of R–CH–X moieties (C-027, X is a non-C or H atom)); Burden BCUT descriptor-molecular eigenvalue based on atomic mass (BEHm7); aromaticity index based on length of conjugated pathway (HOMT); the number of R–CX–X moieties (C-044); the number of phenol/enol/carboxyl OH moieties (O-057); the number of R-SH moieties (S-106). The relevant chemically intuitive in house descriptors were: the number of thiol moieties (SH); the number of sulfur atoms in the molecule (S); the number of hydrogen atoms with intermediate partial charge (BCGM2); the number of carbon atoms of lower partial charge (BCGM4); the number of carbon atoms of intermediate partial charge (BCGM5); the number of tertiary nitrogen atoms (A11); the number of secondary sulfur atoms (A31).
It is clear that some of the descriptors selected from each family encode similar properties, especially those relating to the sulfur moieties in the inhibitors (S-106, SH, S). The other descriptors are difficult to interpret in terms of corrosion mechanisms. They relate mainly to the aromaticity of the inhibitors (nBnz, HOMT), and heteroaromatic properties of the inhibitors (A11, indirectly nR09). These descriptors encode properties related to sulfur and nitrogen binding to metals and the length of conjugated chains in aromatic or more extended molecules, possibly suggesting π–π interactions that would be involved if self-assembly at metal surfaces was important. Thus it is possible that some of the aromatic inhibitors may be forming ordered layers on the surface, or in the case of compounds that resemble thiophenolates, there may be formation of polymeric complexes on the surface as has been reported in the literature.52,53 It is also likely that some inhibitors such as aliphatic thio-containing compounds may be working via another mechanism again. Clearly the mechanism of interactions of small organic molecules with metal surface is complex and largely unknown. These factors, plus the modest size and chemical diversity of the data set, suggests caution in not over-interpreting the models. Currently, the complexity of corrosion and corrosion inhibition for real systems containing commercial alloys, water, salts etc. is sufficiently complex that only machine learning methods like neural networks are feasible for the modelling of corrosion inhibition. However, this capability is at the expense of much lower mechanistic insight compared to computationally intensive physics-based methods like DFT and molecular dynamics.
These models are able to make predictions of the likely corrosion inhibition of new small molecules not yet tested or synthesized. However, care must be taken to ensure these predictions are close to the domain of applicability of the models (the ranges of the molecular descriptors used to generate the models) or the accuracy of prediction will degrade significantly.
Although the data set we have analysed is relatively small for a QSPR modelling study it is, to the authors’ knowledge, the largest yet analysed to determine correlation between molecular characteristics and experimentally measured corrosion inhibition. As such it is of significant interest that correlations with DFT derived properties were not useful, and that the modelling method found other types of molecular descriptors that could model the corrosion efficiency well.
It should be noted that the DFT derived molecular properties (eqn (1)–(5)) were all derived from three DFT calculations: namely EN, EN−1, EN+1. This dependence reduces the richness of the molecular identifiers. The inability of DFT to correlate with corrosion inhibition may be rooted in the disparate length scales between molecular simulation and the macroscopic measurements of corrosion inhibition, and the suitability of the data for comparison. There are also several computational issues that should be considered before drawing conclusions on the suitability of DFT to provide data for corrosion inhibition QSPR models. Firstly, the DFT calculations are very time- and resource consuming so cannot account for the effect that solvent, ions, and the metal surface have on the corrosion inhibitor molecule and vice versa. The adsorption of a corrosion inhibitor molecules which often features N, O, S containing functional groups or heterocyclic functionality, onto a metal/metal oxide surface will likely be accompanied by a redistribution of charge and states, which may be the result of back-bonding from the surface to the corrosion inhibitor, or the formation of a covalent or ionic bond with the surface. Such surface states may shift or fill mid-gap energy levels, affecting the chemical/electrical characteristics of the surface. In addition, the adsorption of a given corrosion inhibitor molecule may not be a simple associative adsorption with the surface; bond breaking/deprotonation may also occur. Thus, calculation of deprotonation energies of the corrosion inhibitor may be warranted, as this will quantify the likelihood of such an event occurring at room temperature. The inclusion of molecular properties such as charge transfer to and from the surface, the direction of such a transfer, post-adsorption changes in work function, and other inhibitor–surface interactions may allow correlations between DFT calculated properties and experimentally determined corrosion inhibition to be identified in the future.
Correlations between molecular properties and attributes and measured inhibition must span length scales from the atomic (10−10 m) to the macroscopic (10−1 m), the size of the test plate used to measure inhibition. Furthermore, the measured property % inhibition as determined by mass loss is a complex average parameter that is influenced by a wide range of parameters that include, surface preparation, oxygen levels, initiation of anode and cathode activity on a surface and the role of microstructure and intermetallics, transition to metastable pitting and then stable pitting, pit chemistry and the development of pit caps and oxide layers with the inhibition having a potential effect on all these properties. Future work could look at refining the experimental measure to reducing the complexity of the processes to contribute to the measure. For example electrochemical measures such an anodic or cathodic current or electrochemical impedance measurements and equivalent circuits could be used, at least as potentially valuable descriptors to relate observed inhibition to the structures of the inhibitors. However, this will involve a relatively large amount of experimental effort for a library of inhibitors.
4. Conclusions
We have shown that, when applied to a larger data set of small organic corrosion inhibitors, the reported correlation between frontier orbital parameters and inhibitor efficiency disappears. We have also shown that it is possible to generate reasonably robust, predictive, and quantitative models of corrosion inhibition using other types of molecular descriptors encoding molecular properties. These models provide a more promising method of predicting the likely effectiveness of new corrosion inhibitors within the domain of the models. Furthermore, they provide a rational basis for design of new inhibitors that may eventually replace toxic chromate corrosion inhibitors and have much less impact on human health and the environment.Acknowledgements
We acknowledge financial support for this work from Boeing Aerospace, and the CSIRO Advanced Materials Transformational Capability Platform.Notes and references
- M. W. Kendig and R. G. Buchheit, Corros., 2003, 59, 379–400 CrossRef CAS.
- R. M. Park, J. F. Bena, L. T. Stayner, R. J. Smith, H. J. Gibb and P. S. J. Lees, Risk Anal., 2004, 24, 1099–1108 CrossRef PubMed.
- C. B. K. Max Costa, Crit. Rev. Toxicol., 2006, 36, 155–163 CrossRef.
- A. E. Hughes, I. S. Cole, T. H. Muster and R. J. Varley, NPG Asia Mater., 2010, 2, 143–151 CrossRef.
- A. Y. Musa, A. B. Mohamad, A. A. H. Kadhum, M. S. Takriff and W. Ahmoda, J. Ind. Eng. Chem., 2012, 18, 551–555 CrossRef CAS PubMed.
- Z. El Adnani, M. McHarfi, M. Sfaira, M. Benzakour, A. T. Benjelloun and M. Ebn Touhami, Corros. Sci., 2013, 68, 223–230 CrossRef CAS PubMed.
- S. John and A. Joseph, Mater. Corros., 2013, 64, 625–632 CrossRef CAS.
- C. M. Goulart, A. Esteves-Souza, C. A. Martinez-Huitle, C. J. F. Rodrigues, M. A. M. Maciel and A. Echevarria, Corros. Sci., 2013, 67, 281–291 CrossRef CAS PubMed.
- T. Le, V. C. Epa, F. R. Burden and D. A. Winkler, Chem. Rev., 2012, 112, 2889–2919 CrossRef CAS PubMed.
- A. S. Barnard, Nat. Nanotechnol., 2009, 4, 332–335 CrossRef CAS PubMed.
- M. J. Zhu, F. Ge, R. L. Zhu, X. Y. Wang and X. Y. Zheng, Chemosphere, 2010, 80, 46–52 CrossRef CAS PubMed.
- F. R. Burden and D. A. Winkler, Chem. Res. Toxicol., 2000, 13, 436–440 CrossRef CAS PubMed.
- C. A. Doose, J. Ranke, F. Stock, U. Bottin-Weber and B. Jastorff, Green Chem., 2004, 6, 259–266 RSC.
- A. Garcia-Lorenzo, E. Tojo, J. Tojo, M. Teijeira, F. J. Rodriguez-Berrocal, M. P. Gonzalez and V. S. Martinez-Zorzano, Green Chem., 2008, 10, 508–516 RSC.
- R. Hernandez-Altamirano, V. Y. Mena-Cervantes, S. Perez-Miranda, F. J. Fernandez, C. A. Flores-Sandoval, V. Barba, H. I. Beltran and L. S. Zamudio-Rivera, Green Chem., 2010, 12, 1036–1048 RSC.
- B. Jastorff, R. Stormann, J. Ranke, K. Molter, F. Stock, B. Oberheitmann, W. Hoffmann, J. Hoffmann, M. Nuchter, B. Ondruschka and J. Filser, Green Chem., 2003, 5, 136–142 RSC.
- D. A. Winkler, Drugs Future, 2004, 29, 1043–1057 CrossRef CAS.
- G. Gece, Corros. Sci., 2008, 50, 2981–2992 CrossRef CAS PubMed.
- T. G. Harvey, S. G. Hardin, A. E. Hughes, T. H. Muster, P. A. White, T. A. Markley, P. A. Corrigan, J. Mardel, S. J. Garcia, J. M. C. Mol and A. M. Glenn, Corros. Sci., 2011, 53, 2184–2190 CrossRef CAS PubMed.
- T. G. Harvey, unpublished work.
- S. H. Hilal, S. W. Karickhoff and L. A. Carreira, Quant. Struct.-Act. Relat., 1995, 14, 348–355 CrossRef CAS.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Revision A.1 edn., Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- R. Todeschini and V. Consonni, Handbook of Molecular Descriptors, Wiley-VCH, Weinheim, 2000 Search PubMed.
- F. R. Burden, M. J. Polley and D. A. Winkler, J. Chem. Inf. Model, 2009, 49, 710–715 CrossRef CAS PubMed.
- D. A. Winkler and F. R. Burden, Mol. Simul., 2000, 24, 243–258 CrossRef CAS.
- D. A. Winkler, F. R. Burden and A. J. R. Watkins, Quant. Struct.-Act. Relat., 1998, 17, 14–19 CrossRef CAS.
- F. R. Burden and D. A. Winkler, J. Med. Chem., 1999, 42, 3183–3187 CrossRef CAS PubMed.
- F. R. Burden and D. A. Winkler, J. Chem. Inf. Comput. Sci., 1999, 39, 236–242 CrossRef CAS.
- F. R. Burden and D. A. Winkler, QSAR Comb. Sci., 2009, 28, 1092–1097 CAS.
- F. R. Burden and D. A. Winkler, QSAR Comb. Sci., 2009, 28, 645–653 CAS.
- D. Winkler, Drug Discovery Today, 2001, 6, 1198–1199 CrossRef.
- D. Winkler, F. Burden and J. Halley, Drugs Future, 2007, 32, 26–26 CrossRef.
- D. A. Winkler, Mol. Biotechnol., 2004, 27, 139–167 CrossRef PubMed.
- F. R. Burden, M. G. Ford, D. C. Whitley and D. A. Winkler, J. Chem. Inf. Comput. Sci., 2000, 40, 1423–1430 CrossRef CAS.
- D. Husmeier, W. D. Penny and S. J. Roberts, Neural Netw., 1999, 12, 677–705 CrossRef.
- D. A. Winkler, Brief. Bioinform., 2002, 3, 73–86 CrossRef CAS.
- L. S. Liebeskind, J. Srogl, C. Savarin and C. Polanco, Pure Appl. Chem., 2002, 74, 115–122 CrossRef CAS.
- P. A. White, unpublished work.
- M. Breedon, M. C. Per, A. S. Barnard and I. Cole, unpublished work.
- F. Bentiss, M. Lebrini, M. Lagrenee, M. Traisnel, A. Elfarouk and H. Vezin, Electrochim. Acta, 2007, 52, 6865–6872 CrossRef CAS PubMed.
- E. E. Ebenso, T. Arslan, F. Kandemirli, N. Caner and I. Love, Int. J. Quantum Chem., 2010, 110, 1003–1018 CrossRef CAS.
- E. E. Ebenso, T. Arslan, F. Kandemirli, I. Love, C. Odretir, M. Saracoglu and S. A. Umoren, Int. J. Quantum Chem., 2010, 110, 2614–2636 CrossRef CAS.
- N. O. Eddy, S. R. Stoyanov and E. E. Ebenso, Int. J. Electrochem. Sci., 2010, 5, 1127–1150 CAS.
- E. H. El Ashry, A. El Nemr, S. A. Essawy and S. Ragab, Prog. Org. Coat., 2008, 61, 11–20 CrossRef CAS PubMed.
- K. F. Khaled, Corros. Sci., 2011, 53, 3457–3465 CrossRef CAS PubMed.
- K. F. Khaled and N. S. Abdel-Shafi, Int. J. Electrochem. Sci., 2011, 6, 4077–4094 CAS.
- K. F. Khaled, K. Babic-Samardzija and N. Hackerman, Electrochim. Acta, 2005, 50, 2515–2520 CrossRef CAS PubMed.
- M. Outirite, M. Lagrenee, M. Lebrini, M. Traisnel, C. Jama, H. Vezin and F. Bentiss, Electrochim. Acta, 2010, 55, 1670–1681 CrossRef CAS PubMed.
- E. Heitz, in Advances in Corrosion Science and Technology, ed. M. G. Fontana and R. W. Staehle, Plenum Press, New York, 1974, ch. 3, pp. 149–243 Search PubMed.
- X. R. Ye and X. Q. Xin, Acta Chim. Sin., 1995, 53, 462–467 CAS.
- X. R. Ye, X. Q. Xin, J. J. Zhu and Z. L. Xue, Appl. Surf. Sci., 1998, 135, 307–317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc42540a |
| This journal is © The Royal Society of Chemistry 2014 |