 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient 2-step biocatalytic strategies for the synthesis of all nor(pseudo)ephedrine isomers†
Torsten
Sehl
a,
Helen C.
Hailes
b,
John M.
Ward
c,
Ulf
Menyes
d,
Martina
Pohl
a and
Dörte
Rother
*a
aIBG-1: Biotechnology, Forschungszentrum Jülich GmbH, Leo-Brandt-Str. 1, 52425 Jülich, Germany. E-mail: do.rother@fz-juelich.de
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H OAJ, UK
cThe Advanced Centre for Biochemical Engineering, Department of Biochemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK
dEnzymicals AG, Walther-Rathenau-Str. 49a, 17489 Greifswald, Germany
First published on 10th April 2014
Abstract
Chiral 1,2-amino alcohols are important building blocks for chemistry and pharmacy. Here, we developed two different biocatalytic 2-step cascades for the synthesis of all four nor(pseudo)ephedrine (N(P)E) stereoisomers. In the first one, the combination of an (R)-selective thiamine diphosphate (ThDP)-dependent carboligase with an (S)- or (R)-selective ω-transaminase resulted in the formation of (1R,2S)-NE or (1R,2R)-NPE in excellent optical purities (ee >99% and de >98%). For the synthesis of (1R,2R)-NPE, space–time yields up to ∼26 g L−1 d−1 have been achieved. Since a highly (S)-selective carboligase is currently not available for this reaction, another strategy was followed to complement the nor(pseudo)ephedrine platform. Here, the combination of an (S)-selective transaminase with an (S)-selective alcohol dehydrogenase yielded (1S,2S)-NPE with an ee >98% and a de >99%. Although lyophilized whole cells are cheap to prepare and were shown to be appropriate for use as biocatalysts, higher optical purities were observed with purified enzymes. These synthetic enzyme cascade reactions render the N(P)E-products accessible from inexpensive, achiral starting materials in only two reaction steps and without the isolation of the reaction intermediates.
Introduction
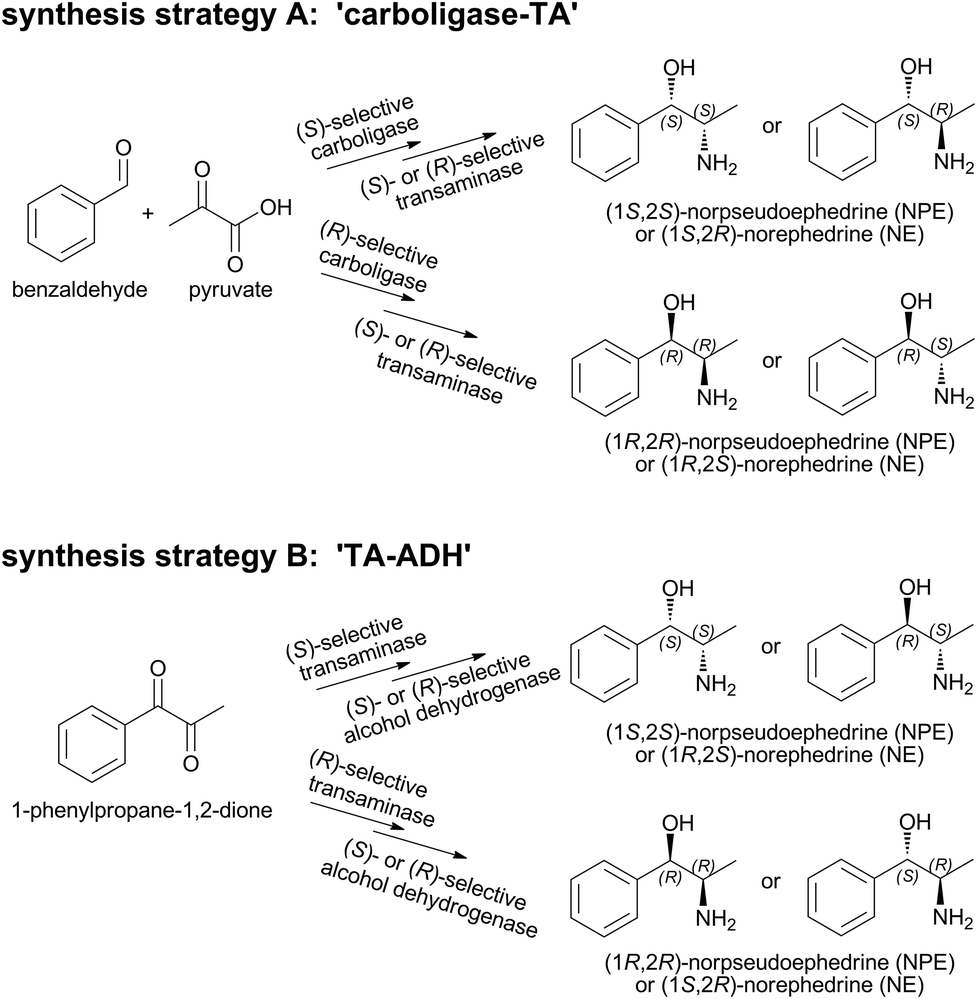
Norpseudoephedrine (NPE) and norephedrine (NE) belong to the amphetamine family of ephedra alkaloids and can be found in plants like Khat (Catha edulis)1,2 and in some Ephedra species.3 In the human body they are known to have a sympathomimetic function4 and act as non-selective adrenergic receptor agonists and norepinephrine re-uptake inhibitors.5 As pharmaceuticals, they have been used to induce mydriasis (dilation of pupils), to stabilize blood pressure, as nasal decongestants, appetite suppressants, and in cold/flu medication.6–9 In most countries N(P)Es are available only on prescription.10 On the German market, cathine ((1S,2S)-NPE) is currently sold for its appetite suppressant function as an active ingredient in ALVALIN®.11,12 In addition to their pharmacological interest, the four N(P)Es stereoisomers are valuable synthons, ligands, and chiral auxiliaries in organic syntheses.13,14 Indeed, a SciFinder® search revealed that N(P)Es have been used as reactants in >5000 different reactions.15So far, >350 different synthetic strategies towards N(P)E stereoisomers have been described in SciFinder®.15 Still, methods for the asymmetric synthesis of nor(pseudo)ephedrines from inexpensive starting materials are rare and require multi-step preparative routes, are based on relatively expensive reagents or lack high enantio- and diastereomeric selectivities.16 Single enzymatic steps17–20 and enzyme cascades21–25 have significant potential in chiral asymmetric synthesis. If a suitable multi-step cascade is performed in one pot, it can be a highly selective, step- and atom-efficient strategy which circumvents a time consuming and expensive isolation of intermediates.26,27
For the synthesis of (1R,2R)-norpseudoephedrine and (1S,2R)-norephedrine, we recently described a 1-pot 2-step enzyme cascade with overall conversions of up to >95%. The combination of a highly (R)-selective carboligase with either an (S)- or (R)-selective ω-transaminase (TA) gave access to these products in high optical purities of ee >99% and de >98%.28
However, synthesis of the two remaining stereoisomers (1S,2S)-NPE and (1S,2R)-NE requires a highly (S)-selective thiamine diphosphate (ThDP)-dependent carboligase in the first reaction step (Scheme 1A), which is currently not available among known wild-type enzymes. Due to our knowledge of the reaction mechanism and factors influencing chemo- and stereoselectivities of ThDP-dependent enzymes, a variant has been designed of the pyruvate decarboxylase from Acetobacter pasteurianus, producing phenylacetylcarbinol (PAC) (S)-selectively for the first time.29
 | ||
| Scheme 1 Two 1-pot 2-step strategies for the synthesis of nor(pseudo)ephedrines combining (A) carboligases and transaminases (TAs) and (B) TAs and alcohol dehydrogenases (ADHs). | ||
Here, we describe the evaluation of this enzyme for the analogous 1-pot 2-step synthesis of (1S,2R)-NE and (1S,2S)-NPE. To avoid a chiral purification step of the resulting nor(pseudo)ephedrine diastereomers, an alternative synthetic strategy (‘TA-ADH’, Scheme 1B) was investigated. Starting from 1-phenylpropane-1,2-dione (1,2-PPDO), in principle all four N(P)E isomers should be accessible in two steps by combining respective stereoselective ω-TAs in the first with alcohol dehydrogenases (ADHs) in the second cascade step. We discuss here advantages and bottlenecks of both strategies.
Synthesis strategy A: carboligase-TA
A-1: (1R,2R)-NPE – high space–time-yields using benzaldehyde emulsions
To date, the biocatalytic 2-step synthesis of (1R,2R)NPE (Scheme 2) was only performed in aqueous buffer with low substrate concentrations (maximum of 20 mM benzaldehyde) with space–time-yields (STY) of ∼2 g L−1 d−1.28 To increase the productivity of this cascade, the enzyme concentration was optimized and its performance in the presence of higher benzaldehyde concentrations was investigated (up to 100 mM benzaldehyde).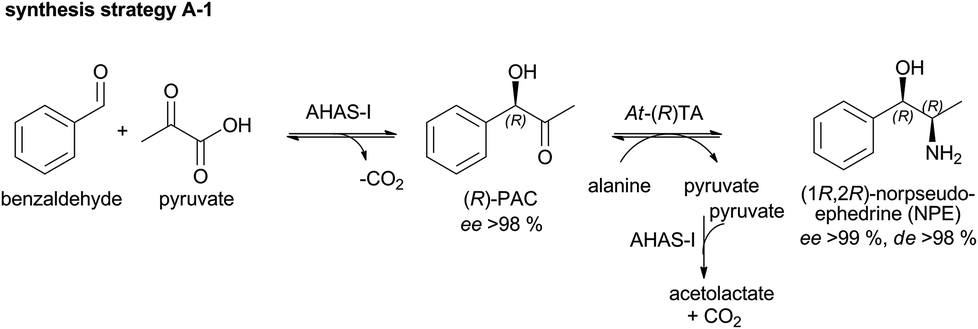 | ||
| Scheme 2 Strategy A-1: combination of the (R)-selective AHAS-I from E. coli and (R)-selective transaminase from A. terreus (At-(R)TA) for the synthesis of (1R,2R)-NPE. | ||
The 2-step cascade, a combination of the AHAS-I from E. coli and the (R)-selective TA from Aspergillus terreus (At-(R)TA) for the synthesis of (1R,2R)-NPE, could be performed in one pot.28 Starting from equimolar concentrations of pyruvate and benzaldehyde (10 mM each) in the carboligation step, the reductive amination could be performed in an optimized manner with a 5-fold excess of D-alanine as the co-substrate. This one pot approach is feasible without addition of further compounds or enzymes, despite the low equilibrium constant of the reductive amination (Keq = 2.31 × 10−3), because AHAS-I removes the co-product pyruvate by converting it to acetolactate and thus shifts the equilibrium to the product side.28 Although the cascade could be performed in a simultaneous mode (both enzymes added simultaneously), the sequential mode (TA added after the AHAS-I reaction was completed) proved to be advantageous due to a lower by-product formation (benzylamine, formed by reductive amination of benzaldehyde).28 In order to further optimize the biocatalytic approach, the minimum amount of enzymes required for a reaction containing 20 mM benzaldehyde, 20 mM pyruvate, and 100 mM D-alanine (see Fig. 1) was determined.
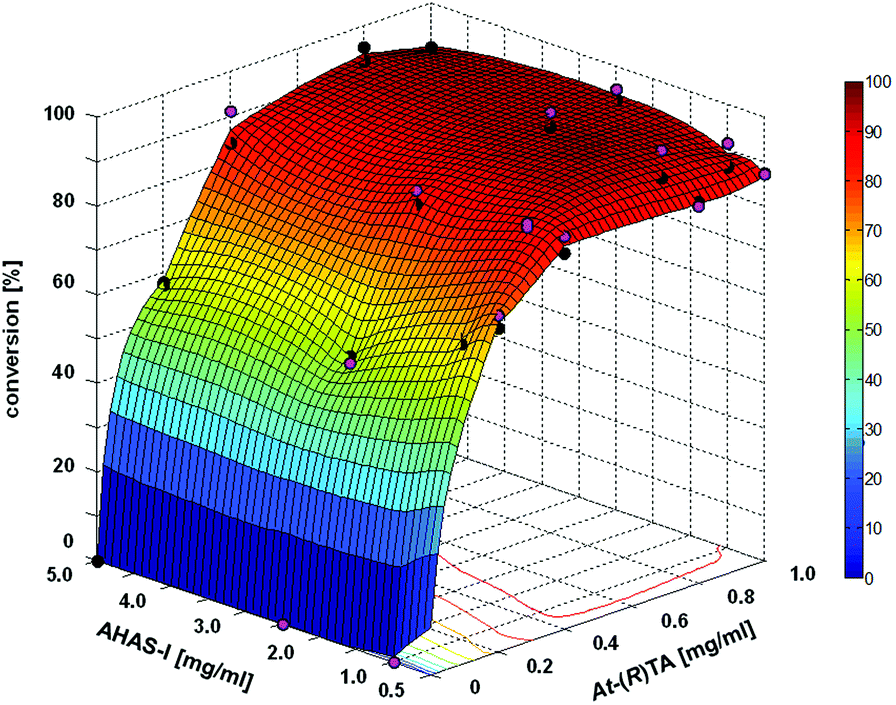 | ||
| Fig. 1 Reaction optimization for the 1-pot 2-step sequential cascade of (1R,2R)-NPE with different concentrations of AHAS-I and At-(R)TA. Reaction parameters: 20 mM benzaldehyde, 20 mM pyruvate and 100 mM D-alanine in 100 mM HEPES (pH 7.5 with 200 μM PLP, 50 μM FAD, 100 μM ThDP, 5 mM MgCl2). Without isolation of the reaction intermediate (R)-PAC, lyophilized At-(R)TA was added 90 min after the addition of AHAS-I (in the given concentrations (pink dots)). The complete reaction was analyzed after another 12 h. | ||
It was found that 0.5 mg mL−1 AHAS-I and 0.4 mg mL−1At-(R)TA were sufficient to achieve 95% conversion of 20 mM benzaldehyde within 13.5 h (Fig. 1). This corresponds to a space–time yield of ∼5 g L−1 d−1 over both reaction steps.
To enhance the reaction efficiency further, the initial benzaldehyde (BA) and pyruvate concentration was increased to 25, 50, 75 and 100 mM but using a constant 5-fold excess of alanine. In line with the optimized reaction parameters, per mmol benzaldehyde 0.025 mg mL−1 AHAS-I (90 min reaction time) and 0.02 mg mL−1At-(R)TA (12 h reaction time) were added.
Although benzaldehyde is not soluble in aqueous buffer >50 mM, in all cases conversions of up to 90% were observed with less than 5% benzylamine by-product formation (Fig. 2). Interestingly, this emulsion system did not significantly influence the reaction performance. In line with these data, finally space–time-yields up to 26 g L−1 d−1 (1R,2R)-NPE were achieved starting from 100 mM benzaldehyde and 90% conversion could be observed within 13.5 h.
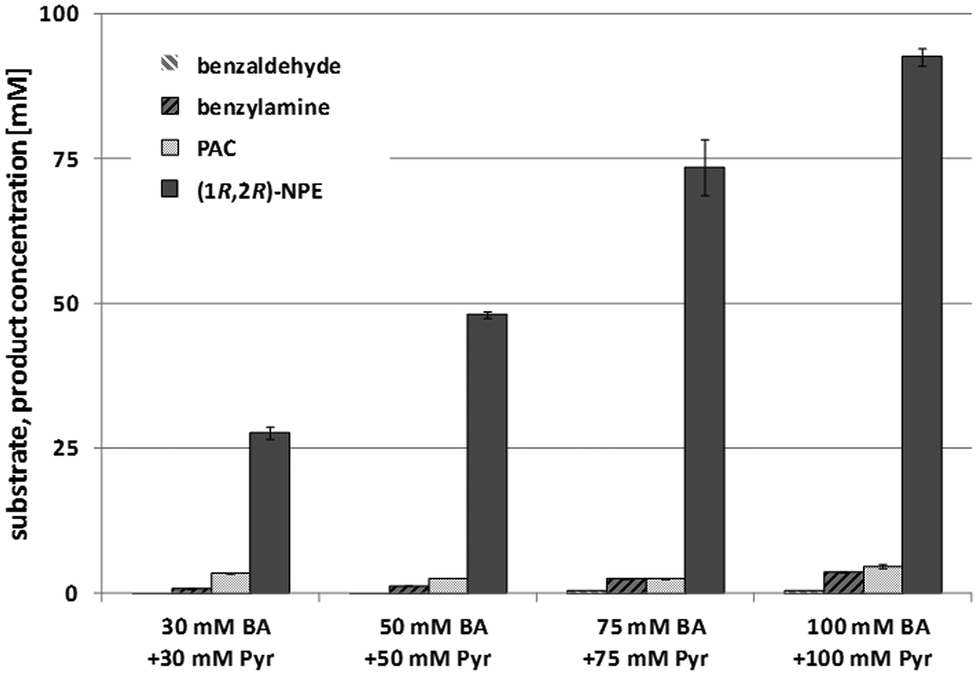 | ||
| Fig. 2 1-Pot 2-step sequential cascade for the synthesis of (1R,2R)-NPE with 25, 50, 75 or 100 mM benzaldehyde (BA) and equimolar concentrations of pyruvate (Pyr). 0.025 mg mL−1 AHAS-I were used per mM benzaldehyde. After 90 min reaction time, 0.02 mg mL−1At-(R)TA and 2.5 mM D-alanine were added per mM initial benzaldehyde concentration. The solution was extracted after a further 12 h reaction time. Reaction parameter: 100 mM HEPES pH 7.5, 200 μM PLP, 50 μM FAD, 100 μM ThDP, 5 mM MgCl2, 25 °C. | ||
A-2: (1R,2S)-NE – application of lyophilized whole cells
The use of whole cells can reduce the catalyst cost by a factor of 10 and is particularly advantageous if side reactions do not occur.30 For an economically feasible large scale application, access to (1R,2S)-NE has been demonstrated by a combination of AHAS-I and Cv-(S)TA (Scheme 3).28 Here, the use of enzymes as lyophilized whole cells without enzyme purification was investigated, which features the benefit of easy handling compared to wet cells (e.g. in terms of weighing out small quantities). | ||
| Scheme 3 Synthetic strategy A-2: combination of the (R)-selective AHAS-I and the (S)-selective transaminase Cv-(S)TA gives access to (1R,2S)-NE. | ||
Concerning the first step, purified AHAS-I shows a high initial rate activity of ∼1.8 U mgprotein−1 for the catalyzed carboligation of benzaldehyde and (decarboxylated) pyruvate, which is within the range of industrially suitable catalysts17 (Fig. 3A). The use of 5 mg mL−1 of lyophilized recombinant E. coli whole cells (LWC) yielded a similar reaction velocity under equivalent reaction conditions (Fig. 3B), which is a consequence of the lower protein amount per mgcatalyst (for comparison of AHAS-I concentrations see SDS-PAGE in ESI Fig. 1†). In both cases a complete conversion of 10 mM benzaldehyde was achieved within ∼0.5 h. This corresponds to a STY of ∼72 g L−1 d−1 and a specific STY (STY gcatalyst−1) for LWC of ∼14 g L−1 d−1 gLWC−1 and for a purified enzyme of ∼144 g L−1 d−1 genzyme−1.
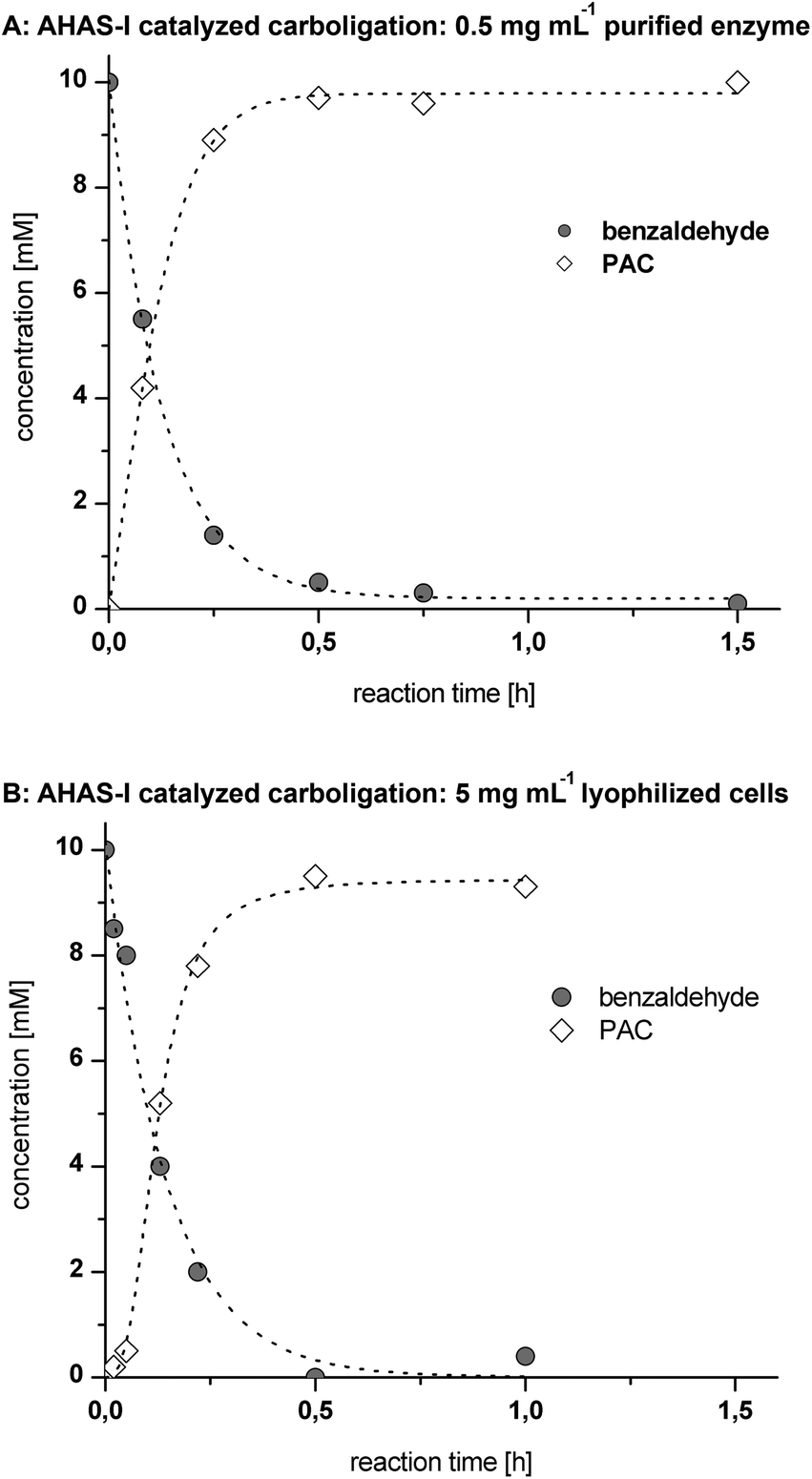 | ||
| Fig. 3 Carboligation of 10 mM benzaldehyde and 10 mM pyruvate catalyzed either by 0.5 mg mL−1 purified AHAS-I (A) or 5 mg mL−1 lyophilized whole cells (B). Each reaction was performed in 100 mM HEPES (pH 7.5, 50 μM FAD, 100 μM ThDP, 5 mM MgCl2) at 25 °C. | ||
Additionally, a comparison of LWC and purified enzymes for the second reductive amination step of (R)-PAC using the Cv-(S)TA was investigated. In a first trial with L-alanine as an amine donor no NPE product formation was observed (data not shown). However, exchange of L-alanine by (S)-α-methylbenzylamine ((S)-α-MBA) in a reaction containing 10 mM (R)-PAC and 5 mg mL−1 LWC resulted in a conversion of 85% (see Fig. 4). Notably, we observed a lag phase of 60 min before product formation started when the reductive amination step was catalyzed with lyophilized whole cells (Fig. 4). This lag phase seems to be caused by membrane required rehydration time of the lyophilized whole cells in the aqueous buffer and could be reduced by a pre-incubation of the LWC in reaction buffer (see ESI chapter 3.1.4†). Still, the reaction rate with 5 mg mL−1 LWC was almost as fast as with 1 mg mL−1 purified enzyme. In both cases a conversion of ∼85% was reached within 3 h, which corresponds to a STY of ∼10 g L−1 d−1. According to this, the calculated specific STY for LWC is ∼2 g L−1 d−1 gLWC−1 and for purified enzyme ∼10 g L−1 d−1 genzyme−1.
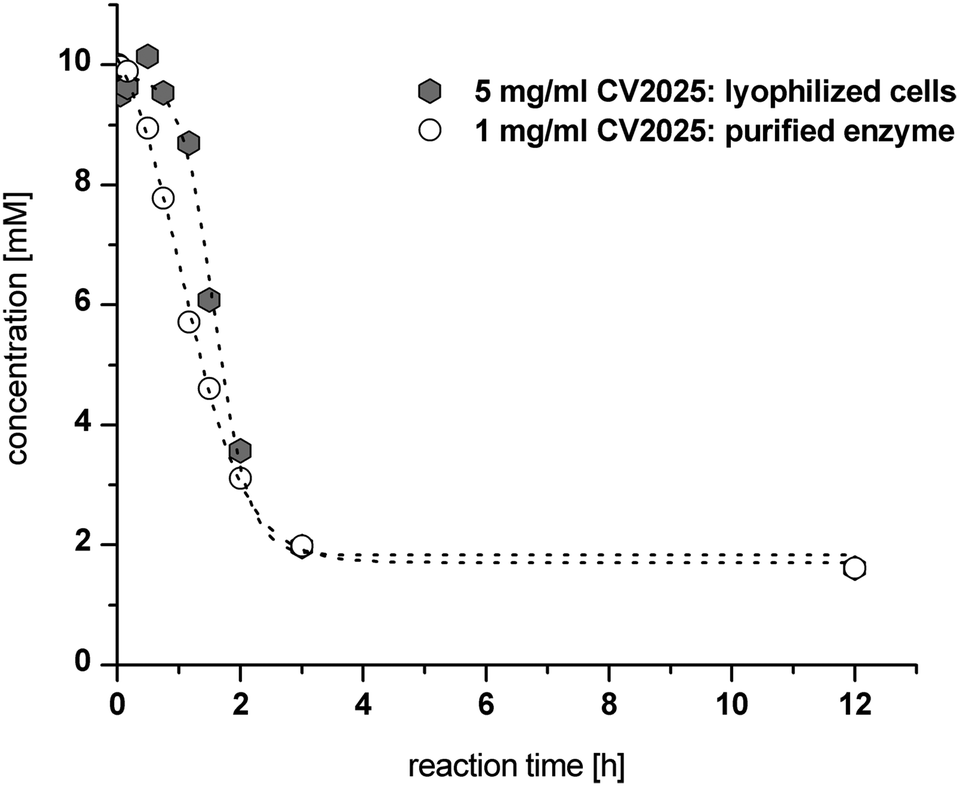 | ||
| Fig. 4 Cv-(S)TA catalyzed reductive amination of 10 mM (R)-PAC in the presence of 10 mM (S)-α-MBA catalyzed by either purified enzyme (1 mg mL−1) or lyophilized cells (5 mg mL−1). | ||
The application of lyophilized whole cells, in general, was found to be suitable for the “carboligase-TA” cascade. This is of special interest for a reaction process, since overall process costs are reduced when enzyme purification can be avoided. Compared to a similar reaction with lyophilized enzymes, the catalyst cost can be significantly lowered by a factor of 10, as was suggested by a general estimation of the catalyst cost by Tufvesson and co-workers.30
A-3, A-4: access to (1S,2R)-NE and (1S,2S)-NPE
The most promising enzyme for (S)-selective carboligation of benzaldehyde and acetaldehyde is ApPDC-E469G.29,31 Although the stereoselectivity for (S)-PAC of 89% ee is not optimal, the 2-step cascade in combination either with an (S)- or (R)-selective ω-TA would result in the formation of (1S,2S)-NPE (Scheme 4; A-3) or (1S,2R)-NE (Scheme 4; A-4) if (S)-PAC is accepted as substrate. | ||
| Scheme 4 Synthesis strategy A-3, A-4: combination of the (S)-selective carboligase ApPDC-E469G with either the (S)-selective TA Cv-(S)TA (A-3) or the (R)-selective TA At-(R)TA (A-4) gives access to (1S,2S)-NPE (A-3) or (1S,2R)-NE (A-4), respectively. | ||
The carboligation step catalyzed by ApPDC-E469G resulted in a final conversion of 95% within 48 h (40 mM benzaldehyde, 400 mM pyruvate, 2.5 mM MgSO4, 100 μM ThDP, 50 mM potassium phosphate, pH 7).32 Under these reaction conditions, the obtained (S)-PAC had an ee of only ∼70%. Its following reductive amination using 10 mM isolated (S)-PAC with 15 mM (R)- or (S)-α-MBA, respectively, and (R)-selective At-(R)TA resulted in the formation of predominantly (1S,2R)-NE with >95% conversion. With the (S)-selective Cv-(S)TA, (1S,2S)-NPE was produced with similar conversions >95%. As expected, both products had a high enantiomeric purity of >99%, but only a low diastereomeric excess of ∼70%. These results highlight that both PAC enantiomers are accepted by both ω-TAs and are further reduced with high stereoselectivity. The missing influence of the chiral vicinal hydroxyl group on the TA reactivity is in line with data for a similar substrate (1,3-dihydroxy-1-phenylpropane-2-one) published earlier.33 To further increase the optical purities of the N(P)E in our synthetic cascade approach, a catalyst with higher stereoselectivity for the (S)-PAC synthesis would be required. Since so far further rational design attempts have been unsuccessful, the combination of ω-TA with oxidoreductases (synthesis strategy B) was subsequently investigated to access these two products in higher optical purities.
Synthesis strategy B: TA–ADH
A novel reaction cascade consisting of a TA in the first step and an ADH in the second step could circumvent the low optical purity in the “carboligase-TA” cascade for the synthesis of (1S,2R)-NE and (1S,2S)-NPE. Using 1-phenylpropane-1,2-dione (1,2-PPDO) as a substrate, the combination of an (S)-selective TA (here Cv-(S)TA) either with the (R)-selective ADH from Ralstonia sp. (RADH) or the (S)-selective ADH from Lactobacillus brevis (LbADH) could give access to (1R,2S)-NE and (1S,2S)-NPE (strategy B-1; Scheme 5). Since the enzymes for both steps are highly selective for aryl-aliphatic substrates, as was earlier demonstrated for oxidoreductases34 and ω-TAs28 the products should be accessible in high optical purity. To complete the strategies towards all possible stereoisomers of N(P)E with high stereoselectivities, the access to (1S,2R)-NE and (1R,2R)-NPE was investigated by a combination of the (R)-selective At-(R)TA with LbADH and RADH, respectively (strategy B-2; Scheme 5). Finally, the application of different enzyme preparations (lyophilized whole cells, purified enzymes or crude cell extract) was compared.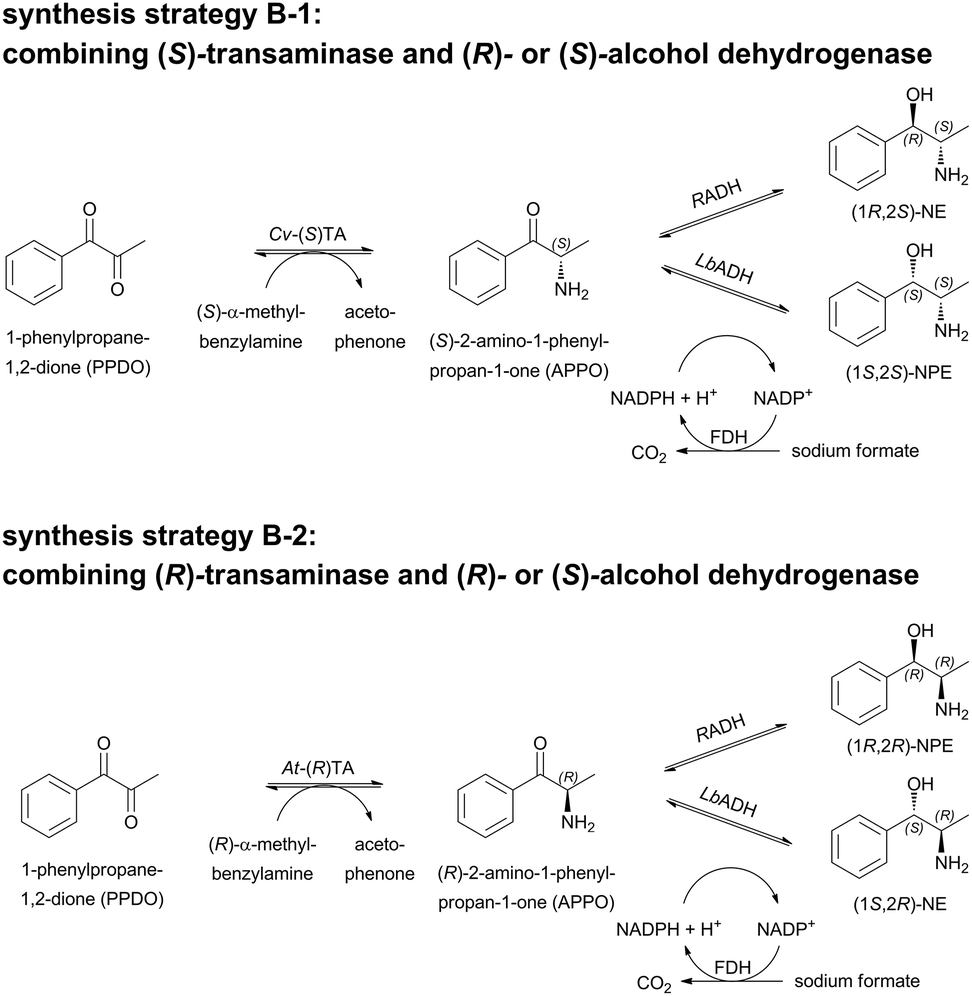 | ||
| Scheme 5 Strategy B: enzymatic 1-pot 2-step reaction for the synthesis of all nor(pseudo)ephedrines combining either an (S)-selective (Cv-(S)TA, strategy B-1) or an (R)-selective transaminase (At-(R)TA, strategy B-2) in the first step and an (R)- (RADH) or (S)-selective (LbADH) alcohol dehydrogenase (ADH) in the second step. | ||
In order to determine optimal reaction conditions for the reductive amination of 1,2-PPDO, reactions with lyophilized whole cells (LWC) containing Cv-(S)TA and different concentrations of the amine donor (S)-α-MBA were investigated and applied to the reductive amination with At-(R)TA (see ESI†). A concentration of 15 mM (S)-α-MBA was sufficient to achieve full conversion of 10 mM 1,2-PPDO with Cv-(S)TA in 48 h. With higher concentrations of the amine donor (S)-α-MBA the acetophenone concentration (co-product formed upon deamination of (S)-α-MBA) did not increase to more than 10 mM, indicating that the theoretically possible di-amination of the diketone 1,2-PPDO did not occur. This is in accordance with previous results in which the reductive amination of aryl-aliphatic ketones can only be achieved if the aliphatic group is not larger than an ethyl group.35 The initial rate activity for the reductive amination of 1,2-PPDO towards (S)-APPO was ∼0.005 U mgLWC−1 with LWC containing overexpressed Cv-(S)TA or ∼0.008 U mgprotein−1 with purified Cv-(S)TA (see ESI;† 1 U corresponds to the amount of enzyme that catalyzes the conversion of 1 μmol min−1 1,2-PPDO under standard conditions). In contrast to this, in a reaction with E. coli crude cell extract containing At-(R)TA (Scheme 5 B-1) a significantly higher activity of ∼0.1 U mgprotein−1 was measured for the synthesis of (R)-APPO. Here, in all cases a complete conversion of the diketone substrate was observed in only 3 h (see ESI†).
The subsequent reduction reaction of the intermediate product APPO can in principle be performed in the same pot without intermediate isolation. The required cofactor NADPH (0.5 mM) was regenerated by the addition of formate dehydrogenase (FDH) from Pseudomonas sp. and sodium formate, based upon a method described previously.36 When the cascade was performed in the sequential mode without quenching the TA reaction, 1-phenylpropane-1,2-diol was detected as a by-product. This was most likely due to the reversibility of the TA reaction and an equilibrium that favoured the di-reduction of PPDO by ADH. To suppress the diol formation, inactivation of the TA after the first reaction step was investigated. Either a pH-shift (pH 7.5 to pH 2 with 20% (v/v) HCl – and re-titration to pH 7.5 with 1 M NaOH) or an ultrafiltration step to remove the TA was tested. Using inactivation by pH-shift, the combination Cv-(S)TA/LbADH as purified enzymes gave an overall conversion of 80% after 52 h. With this biocatalytic cascade the product (1S,2S)-NPE was accessible for the first time with high optical purity: de >99% and ee >98% (Table 1; entry #4). When ultrafiltration was used to remove the TA for the combination of Cv-(S)TA/LbADH with purified enzymes, the 2-step conversion was significantly lower (57% after 60 h), but ee and de remained equally high (Table 1; entry #2 compared to #4). The same trend was found for the combination of Cv-(S)TA/RADH with purified enzymes, where the product (1R,2S)-NE was accessible with ee >99% and de >95% (Table 1; entry #3). Here, the 2-step cascade with an ultrafiltration step after the TA reaction resulted in an overall conversion of 55%, whereas 82% conversion to (1R,2S)-NE was achieved using the pH-shift for TA inactivation (Table 1; entry #3 compared to #5). Possible explanations for the reduced yields upon ultrafiltration might be that centrifugation was carried out in plastic ware (all other steps were performed in closed glass vials) and evaporation and adsorption of the intermediate might lower the final yield significantly. However, this was not investigated in detail because the cheaper pH-shift method solved the problem and even higher ee- and de-values (ee 99%, de 97%).
| Entry | TA reactiona | ADH reactionc | Overall conversion & total reaction timed | Specific STYe [g L−1 d−1 gcat−1] | Product ratio [%] and optical purity of major isomer (bold) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Type | Terminationb | Catalyst | Type | (1S,2S)-NPE | (1R,2R)-NPE | (1S,2R)-NE | (1R,2S)-NE | Optical purity | |||||
| ee | de or dr | |||||||||||||
| TA: transaminase, ADH: alcohol dehydrogenase, Purif.: purified, CCE: crude cell extract, LWC: lyophilized whole cells, NE: norephedrine, NPE: norpseudoephedrine – time dependent reaction curves and reaction analytics can be found in the ESI.a Reaction conditions for reductive amination were carried out as indicated with purified enzyme (1 mgprotein mL−1), CCE (1 mgprotein mL−1) or LWC (10 mgLWC mL−1) of the respective transaminases (At-(R)TA or Cv-(S)TA) in 100 mM HEPES (pH 7.5), 200 μM PLP, with ∼10 mM 1,2-PPDO (see ESI) and 15 mM (R)- or respective (S)-α-MBA at a reaction temperature of 21 °C. As indicated (by joined rows: #2, 3 – #4, 5 – #6, 7 – #8, 9 and #10, 11), the reaction solutions were split for the subsequent oxidoreduction step.b Reactions were either terminated by ultrafiltration (membrane cut-off: 10 kDa) and pH-shift (titrated with 20% (v/v) HCl to pH 2 and then re-titrated with 10 M NaOH to pH 7.5) or not terminated.c Reaction conditions for reductive hydrogenation: 0.5 mM NADP+, 150 mM sodium formate, 10 μL mL−1 FDH. Either purified enzyme (1 mgprotein mL−1) or lyophilized whole cells (10 mgLWC mL−1) of the respective alcohol dehydrogenases (LbADH or RADH) were added to this reaction solution and incubated at 21 °C.d Overall conversion (sum of N(P)E related to the initial substrate concentration) and the total reaction time are given for the complete 2-step reaction.e Specific space–time-yields (STY) are calculated from the overall conversion valuesd, the reaction timesd and the amount of catalyst useda. | ||||||||||||||
| #1 | Cv-(S)TA | Purif. | — | LbADH | Purif. | 0% | 48 h | — | — | — | — | — | — | — |
| #2 | Cv-(S)TA | Purif. | Filtration | LbADH | Purif. | 57% | 60 h | 0.17 | 99.3 | 0.7 | — | — | >98% | de >99% |
| #3 | Filtration | RADH | Purif. | 55% | 60 h | 0.17 | — | 2.4 | — | 97.6 | >99% | de ∼95% | ||
| #4 | Cv-(S)TA | Purif. | pH-shift | LbADH | Purif. | 80% | 52 h | 0.28 | 99.4 | 0.6 | — | — | >98% | de >99% |
| #5 | pH-shift | RADH | Purif. | 82% | 40 h | 0.37 | — | 1.5 | — | 98.5 | >99% | de 97% | ||
| #6 | At-(R)TA | CCE | pH-shift | LbADH | Purif. | 40% | 36 h | 0.20 | 79.9 | — | 20.1 | — | >99% | de ∼60% |
| #7 | pH-shift | RADH | Purif. | >95% | 8 h | 2.15 | — | 77.4 | — | 22.6 | >99% | de ∼55% | ||
| #8 | Cv-(S)TA | LWC | pH-shift | LbADH | LWC | 62% | 36 h | 0.03 | 92.0 | 3.0 | 5.0 | — | ∼92% | de ∼90% |
| #9 | pH-shift | RADH | LWC | 67% | 27 h | 0.05 | 2.1 | 17.6 | — | 80.3 | >99% | dr ∼8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 :2 |
||
| #10 | At-(R)TA | CCE | pH-shift | LbADH | LWC | 77% | 30 h | 0.08 | 77.3 | 2.0 | 20.7 | — | ∼95% | de ∼80% |
| #11 | pH-shift | RADH | LWC | 93% | 11 h | 0.28 | — | 75.2 | 0.9 | 23.9 | >99% | dr ∼7.5:2.5 |
||
Compared to the excellent optical purity of (1S,2S)-NPE obtained in the 2-step reaction with purified enzymes (see above), the biotransformations using lyophilized whole cells yielded products with decreased optical purities (Table 1; entry #8, 9): Cv-(S)TA/LbADH ((1S,2S)-NPE: ee >92%, de 90%) and Cv-(S)TA/RADH ((1R,2S)-NE: ee >99%, dr ∼8:2). Here, a decrease in optical purity might be caused by the isomerization of (S)-APPO, induced by other E. coli enzymes. Although it was not possible to measure the ee for the intermediate product APPO, this isomerization could lead to a decrease of the de for the N(P)E products even if both cascade enzymes (ADH and TA) are highly selective.
Similar results were obtained when a crude cell extract containing At-(S)TA was used for the reductive amination step. In combination with RADH (purified enzyme) high conversions (>90%) but rather low optical purities ((1R,2R)-NPE: ee >99%, de >55%) were detected (Table 1; entry #7). Moreover, the combination At-(R)TA/LbADH did not result in (1S,2R)-NE as the major product, but gave (1S,2S)-NPE (Table 1; entry #6, 10). As mentioned for the cascades with Cv-(S)TA lyophilized whole cells, an isomerization of APPO could lead to a decrease in de values even if the enzymes applied are highly selective.
As a consequence, purified enzymes rather than whole cells or crude cell extracts are the method of choice for the TA/ADH reaction cascade. As shown for the combination Cv-(S)TA/LbADH, (1S,2S)-NPE is accessible in high optical purities (>99% de, >98% ee) when purified enzymes are used in both reaction steps (Table 1; entry #4).
Conclusions
In summary, two biocatalytic cascade strategies have been developed for the synthesis of all four phenylpropanolamine stereoisomers and the use of inexpensive whole cells for the reaction steps has been investigated. In the “carboligase-TA” strategy, a combination of the (R)-selective AHAS-I from E. coli either with an (S)- or a (R)-selective ω-TA gave access to (1R,2S)-NE and (1R,2R)-NPE in high optical purities (ee >99% and de >98%). As a proof-of-principle we demonstrated that the synthesis of (1R,2R)-NPE could be performed with substrate concentrations of up to 100 mM by the combination AHAS-I/At-(R)TA. Space–time yields up to ∼26 g L−1 d−1 were achievable. Moreover, the application of LWC is possible, but leads to a reduction in specific STY by a factor of 10 for the C–C coupling reactions and by a factor of 5 for the reductive transaminations. Since a highly (S)-selective enzyme for the synthesis of the intermediate (S)-PAC is currently not available, the products (1S,2R)-NE and (1S,2S)-NPE were only accessible in moderate optical purities using this strategy.An alternative 2-step synthesis strategy combining ω-TA with ADHs was evaluated to gain access to all N(P)E isomers in higher optical purities. Indeed, the isomer (1S,2S)-NPE, also known as cathine, was synthesized enzymatically in high optical purities. Here, the combination of the (S)-selective TA Cv-(S)TA with the (S)-selective alcohol dehydrogenase LbADH gave (1S,2S)-NPE with an ee >98% and a de >99% when purified enzymes were used. This novel biocatalytic reaction cascade can be performed in one pot without isolation of intermediates. However, a deactivation of the ω-TA prior to the reductive hydrogenation was required. Although it was demonstrated that this reaction could be carried out with cheaply produced lyophilized whole cells, there are two major drawbacks. On the one hand, the specific STYs are significantly lower with lyophilized whole cells, which partially negate the 10-fold lower catalyst production costs of cells. On the other hand, the optical purity of the product was higher with purified enzymes. Since chiral product purification methods might dramatically increase production costs, the use of purified enzymes can be beneficial if the additional enzyme purification costs are below downstream processing costs. For industrial applications immobilization of enzymes might be a method to decrease production costs (e.g. in terms of downstream processing, recyclability) and in some cases immobilization results in an increased catalyst stability and/or activity.37–42
In general, it has been demonstrated that enzymes from different toolboxes can be efficiently combined yielding all stereoisomers of desired N(P)E. Here, the “TA-ADH” reaction cascade was developed as an alternative to the “carboligase-TA” cascade giving access to optically pure (1S,2S)-NPE in only two biocatalytic steps.
Acknowledgements
This work was supported by the CLIB Graduate Cluster Industrial Biotechnology of the Heinrich-Heine-University Düsseldorf.Notes and references
- J. M. Hagel, R. Krizevski, K. Kilpatrick, Y. Sitrit, F. Marsolais, E. Lewinsohn and P. J. Facchini, Genet. Mol. Biol., 2011, 34, 640–646 CAS.
- O. Wolfes, Arch. Pharm., 1930, 268, 81–83 CrossRef CAS.
- R. Krizevski, E. Bar, O. Shalit, A. Levy, J. M. Hagel, K. Kilpatrick, F. Marsolais, P. J. Facchini, S. Ben-Shabat and Y. Sitrit, Phytochemistry, 2012, 81, 71–79 CrossRef CAS PubMed.
- Micromedex 2.0®, Micromedex® Healthcare Series [Internet database], Greenwood Village, Colo: Thomson Reuters (Healthcare) Inc., Updated periodically Search PubMed.
- B. B. Hoffman and R. J. Lefkowitz, in The pharmacological basis of therapeutics, The McGraw-Hill Companies Inc., 1996, pp. 222–224 Search PubMed.
- L. Lasagna, Phenylpropanolamine: A review, John Wiley & Sons Inc., 1988 Search PubMed.
- J. P. Morgan, Phenylpropanolamine: A critical analysis of reported adverse drug reactions and overdosage, Jack K Burgess, 1986 Search PubMed.
- M. Weintraub, in Phenylpropanolamine: Risks, Benefits and Controversies, Greenwood Publishing Group, Inc., 5th edn, 1985, pp. 53–79 Search PubMed.
- W. N. Kernan, C. M. Viscoli, L. M. Brass, J. P. Broderick, T. Brott, E. Feldmann, L. B. Morgenstern, J. L. Wilterdink and R. I. Horwitz, N. Engl. J. Med., 2000, 343, 1826–1832 CrossRef CAS PubMed.
- M. Yakoot, J. Pharmacol. Pharmacother., 2012, 3, 4–6 CrossRef CAS PubMed.
- Aponet Arzneimitteldatenbank – Arzneimitteldetails zu “ALVALIN”, http://www.aponet.de/wissen/arzneimitteldatenbank/suchergebnis/arzneimitteldetails/alvalin-40mg-g_3345428700/dosierung.html.
- G. Laux and O. Dietmaier, in Psychopharmaka, Springer, 2013, pp. 189–195 Search PubMed.
- L. Yan, Z. Diansong, S. S. Ferguson, P. Dorff, T. R. Simpson and S. W. Grimm, Xenobiotica, 2010, 40, 721–729 CrossRef PubMed.
- D. G. Allen, N. M. Aston, N. Trivedi and C. D. Edlin, Google Patents, WO2004024728 A3, 2007 Search PubMed.
- American Chemical Society – SciFinder®, https://scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf.
- H. K. Lee, S. Kang and E. B. Choi, J. Org. Chem., 2012, 77, 5454–5460 CrossRef CAS PubMed.
- H. Gröger, Y. Asano and O. May, in Enzyme Catalysis in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 1–42 Search PubMed.
- J. Wang and W. Lu, in Chiral Drugs, John Wiley & Sons, Inc., 2011, pp. 77–136 Search PubMed.
- K. Faber, in Biotransformations in Organic Chemistry, Springer, Berlin, Heidelberg, 2010, ch. 1, pp. 1–30 Search PubMed.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622–640 CrossRef CAS.
- S. F. Mayer, W. Kroutil and K. Faber, Chem. Soc. Rev., 2001, 30, 332–339 RSC.
- J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Org. Chem. Biol., 2011, 15, 249–256 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- F. Lopez-Gallego and C. Schmidt-Dannert, Curr. Org. Chem. Biol., 2010, 14, 174–183 CrossRef CAS PubMed.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 6772–6775 CrossRef CAS PubMed.
- D. Rother, G. Kolter, T. Gerhards, C. L. Berthold, E. Gauchenova, M. Knoll, J. Pleiss, M. Müller, G. Schneider and M. Pohl, ChemCatChem, 2011, 3, 1587–1596 CrossRef CAS.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2010, 15, 266–274 CrossRef.
- T. Gerhards, U. Mackfeld, M. Bocola, E. von Lieres, W. Wiechert, M. Pohl and D. Rother, Adv. Synth. Catal., 2012, 354, 2805–2820 CrossRef CAS PubMed.
- Á. Baraibar, E. Lieres, W. Wiechert, M. Pohl and D. Rother, Top. Catal., 2013, 1–11, DOI:10.1007/s11244-11013-10194-z.
- K. Smithies, M. E. B. Smith, U. Kaulmann, J. L. Galman, J. M. Ward and H. C. Hailes, Tetrahedron: Asymmetry, 2009, 20, 570–574 CrossRef CAS PubMed.
- J. Kulig, R. C. Simon, C. A. Rose, S. M. Husain, M. Hackh, S. Lüdeke, K. Zeitler, W. Kroutil, M. Pohl and D. Rother, Catal. Sci. Technol., 2012, 2, 1580–1589 CAS.
- T. Sehl, R. C. Simon, H. C. Hailes, J. M. Ward, U. Schell, M. Pohl and D. Rother, J. Biotechnol., 2012, 159, 188–194 CrossRef CAS PubMed.
- D. Kihumbu, T. Stillger, W. Hummel and A. Liese, Tetrahedron: Asymmetry, 2002, 13, 1069–1072 CrossRef CAS.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS PubMed.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS PubMed.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019–1032 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst preparation, reaction details, reaction analytics and configuration determination. See DOI: 10.1039/c4gc00100a |
| This journal is © The Royal Society of Chemistry 2014 |