 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of some salts on H2O as probed by a thermodynamic signature of glycerol: towards understanding the Hofmeister effects (VII)†
Yoshikata
Koga
*ab and
Peter
Westh
c
aDepartment of Chemistry, The University of British Columbia, Vancouver, B.C., Canada V6T 1Z1. E-mail: koga@chem.ubc.ca
bSuitekijuku, Vancouver, B.C., Canada V6R 2P5
cNSM Research Unit for Functional Biomaterials, Roskilde University, Roskilde, Denmark DK-4000
First published on 25th October 2013
Abstract
The generality of the Hofmeister effects has been questioned of late, and doubts have been cast over their importance in understanding the specific ion effects on the chemistry and physics of biopolymers in aqueous solutions. Recent experimental evidence from modern non-linear spectroscopies points mostly to the direct interaction between the ion and the biopolymer in question that is more important for understanding the Hofmeister effects. On the other hand, our own contribution by higher order thermodynamical studies indicated that the effects of ions on H2O itself may not be denied all together. Namely, we devised a methodology whereby the effect of an ion on H2O is characterized by two orthogonal indices, hydrophobicity and hydrophilicity, by using a third order thermodynamic signature of hydrophobic 1-propanol (1P) as a probe, the 1P-probing methodology. The results indicated that the common anion ranking could be understood in terms of two indices, hydrophobicity and hydrophilicity of an individual ion. In the present work, we make an attempt at probing the effects of the same ions on H2O by a typical hydrophile, glycerol (abbreviated as Gly in this article). Compared with the results of the 1P-probing methodology, we seek to determine how hydrophiles would react to the subtle modification of H2O caused by the presence of an ion, since biopolymers are large amphiphiles with hydrophobic and hydrophilic surfaces. The results indicate that the Gly-probe is much less sensitive than the 1P-probe. We suggest therefore that it is the hydrophobic moieties of biopolymers that mainly give more conspicuous response to the modified H2O by the presence of an ion.
Introduction
Ions and non-electrolyte solutes modify the molecular organization of H2O in a specific manner. As one of their manifestations, they show marked differences in their solvent properties when used as mixed solvents. This was recognized back in 1887 by F. Hofmeister.1–3 He ranked the effects of ions in the order from what reduces the solubility of lysozyme in aqueous solutions to what promotes it. Since then, almost the same ranking seems to apply to a large number of physical/chemical processes in aqueous solutions of biopolymers or colloids, particularly for anions. The left side of the ranking was named “kosmotropes” and the right “chaotropes” with Cl− at about the null position.4 Thus, at the zero-th approximation, it was generally regarded to be the effect of each ion on H2O that dictates the overall properties of the ternary systems. More recent investigations, however, tend to point to direct ion–biopolymer interactions that are more important for the ion-specific effects. Indeed, modern non-linear higher order spectroscopic studies suggested that the bulk H2O away from hydration shells of common ions was left unperturbed.5–11 Hence, the Hofmeister effects must be due to direct interactions between the specific ion and the biopolymer in question. Furthermore, the reversals of the Hofmeister ranking have been observed by modifying the end groups of the biopolymer,12 or by changing the solution compositions.13–15 With these the Hofmeister effects may become non-existent. The close relation between the Hofmeister series of biopolymers and the lyotropic series of colloids has been long noted. Lyklema pointed out in analogy with colloid science that the Hofmeister series ought to be re-examined by taking into account the surface conditions, hydrophobicity or hydrophilicity, of biopolymers in question.41 Meanwhile, Levin et al. claimed to have developed a theory of the surface density profile that could explain a variety of experimental results with a single adjustable parameter, and that could finally shed light on a century old enigma, the Hofmeister series.42Nonetheless, the effects of ions and non-electrolyte solutes on H2O are interesting in their own right and important for fundamental investigations of the multicomponent aqueous solutions. We have recently devised what we call the 1-propanol (1P) probing methodology that was detailed elsewhere.16,17 By applying it to aqueous solutions, we were able to characterize the effects of solutes, non-electrolytes and individual ions on H2O using a pair of coordinates, hydrophobicity, a, and hydrophilicity, b, and thus to characterize the effect of a solute on a two-dimensional map with H2O at the origin. For an individual species the former relates to its hydration number, nH, and the latter to its effect on the degree of S–V cross fluctuation density (proportional to thermal expansivity) of the entire bulk of the solution.18–21 Using this methodology, we found that there are five distinct classes of the effects of a solute on H2O. In particular, the results of a series of studies on general ions by this methodology indicated that kosmotropes all belong either to “hydration centers” or “hydrophobes”, both being interpreted as forming hydration shells around them, while chaotropes were found to be all “hydrophiles” (see below). Furthermore, the anion Hofmeister ranking matched the decreasing order of the distance from the origin for “hydration centers” and “hydrophobes” and then the increasing order of the distance for “hydrophiles” with the null point being H2O itself. Cl−, which is normally regarded as the null point, was found to belong to the “hydration center” and to be very close to the origin. According to our studies,16,17 “hydration centers” were understood to form hydration shells but the bulk H2O away from hydration shells is left unperturbed, while “hydrophobes” also form hydration shells with somewhat enhanced hydrogen bonding within them with concomitant reduction of the hydrogen bond probability of bulk H2O.20,21 “Hydrophiles”, on the other hand, form hydrogen bonds directly with the hydrogen bond network of H2O and retard the degree of fluctuation inherent in pure H2O.18,22,23 Thus, we suggested that the effects of ions on H2O must remain important in understanding the Hofmeister effects. Of course, the individuality of chemical and physical properties of biopolymers is not unimportant. After all, they are large amphiphiles with hydrophobic and hydrophilic moieties. It would therefore be interesting to investigate how a hydrophobe and a hydrophile react to the modification of H2O caused by the presence of an individual ion. The probe 1-propanol (1P) we have used so far is a typical “hydrophobe” in our classification.16,17 The present work shows how a hydrophile, glycerol (abbreviated as Gly in this paper), would react to the modification of bulk H2O caused by the presence of a specific ion following the earlier preliminary study.24
In dealing with aqueous solutions, particular consideration must be given to the composition. We earlier realized20–22 that the solution properties are crucially dependent on the composition in general for aqueous solutions. We found that the aqueous solution generally consists of three distinctive regions, in each of which the mixing scheme (MS), the molecular level scenario of mixing, is qualitatively different from those of other regions. In the H2O-rich region, H2O is modified somewhat depending on the nature of the solute (the details of which were instrumental in classifying the solute into the five classes mentioned above)16,17 but the basic integrity of liquid H2O is retained inasmuch as the hydrogen bond network is connected fleetingly and yet permanently throughout the bulk. H2O is here understood as a highly fluctuating hydrogen bonded assembly and yet hydrogen bonds are bond-percolated.20,21,25,26 In the solute-rich region, the solute molecules tend to cluster together as in the pure state and H2O interacts with such clusters as a single gas-like molecule. In the intermediate region, two kinds of clusters, one rich in H2O and the other in solute, physically mix together. We name these three distinct mixing schemes Mixing Schemes I, II and III from the H2O-rich end. The boundaries between the adjacent MSs are apparent from the anomalous behaviour of the third derivative thermodynamic quantity.20–22 In special cases, the boundary could appear as liquid–liquid phase separation between MS I and MS II or precipitation of a solute at the MS II and MS III boundary.
In the original experiment conducted by Hofmeister,1,3 the first cloud points with 2 wt% lysozyme were determined in terms of the salt composition. They occurred at the mole fraction of 0.056, 0.03, 0.061 and 0.09, respectively, for the Na-salts of SO42−, OAc−, Cl−, and ClO3−. From the description in this paper, it is not clear whether the first cloud point is phase separation or precipitation. We interpret his first cloud point as corresponding to the MS I and II boundary for safety, and we limit our attention to MS I of the multi-component aqueous solutions.
As detailed earlier,16,17 the methodology we use is applicable only to the limited H2O-rich region, MS I. This is based on our earlier findings that within this limited H2O-rich region, MS I, the effects of ions are additive and that the effects of hydrophobic and hydrophilic moieties of amphiphiles are also additive. Similarly, for a multi-component system the effects of each solute are additive as long as the total mole fraction is small enough so that a body of liquid H2O maintains its integrity.16,17
Here, following the previous Gly-probing study for Na-salts of some anions,24 we apply it to Cl-salts of NH4+, (CH3)4N+ (TMA+) and in addition NaCH3COO (Na+OAc−). The latter was included, since we investigated recently how OAc− works as a hydrophobe.16,27
The details of the probing methodology were described elsewhere.16,17 Very briefly, one of the thermodynamic signatures, HEBB (defined below), for the probing component B in the ternary aqueous solution of B and the test sample S is determined as a function of the mole fraction of B, xB, at a fixed initial mole fraction of S, x0S. HEBB shows the xB-dependence pattern unique to the nature of B. For hydrophobic B, it displays a peak type and for a hydrophilic B a bend type anomaly reflecting a qualitative change in the molecular organization of H2O. (See Fig. 5 in the Appendix.) The peak top or the bend point that we name point X is where the integrity of liquid H2O is lost due to the presence of B at the value of xB. We then observe how the HEBB pattern changes as S is added while the characteristic pattern of HEBB is retained. The induced changes, particularly those of the anomalous point, X, are indexed in two orthogonal directions in the graph of HEBBvs. xB. Thus, the B-probing methodology is applicable only up to this mole fraction. The rate of westward shift (to the negative direction of the xB-axis) of point X per unit increase in x0S is defined as hydrophobicity, a. That of the southward shift (to the negative direction of HEBB-axis) is defined as hydrophilicity, b. The shifts in both directions are found generally to be linear to x0S. By trying out a number of typical hydrophobes and hydrophiles for S, we catalogued the induced changes. Thus, we have a way to characterize the effect of an unknown solute S on H2O using a pair of indices, a and b, and to display it in a two-dimensional map with H2O defining its origin.
From the 1P-probing methodology, we drew the following conclusions for each ion studied here: Na+, NH4+, and Cl− are the “hydration centers” with the hydration number nH, 5.2, 1 ± 1, and 2.3 ± 0.6, respectively, leaving the bulk H2O away from hydration shells unperturbed.16,17 The CH3− side of OAc– is a hydrophobe with the total hydration number 3.7 ± 0.7 and reducing progressively the hydrogen bond probability of bulk H2O away from hydration shells. One out of 3.7 H2O molecules hydrates the COO− side of the ion as a hydration center without affecting the bulk H2O.16,27 TMA+ was found to act as a hydrophile which forms hydrogen bonds directly with the hydrogen bond network of H2O and to pin down the fluctuation inherent in liquid H2O.16,28
As discussed in the Appendix, the Gly-probe has an intrinsic disadvantage in comparison with the 1P-probe. Namely, HE1P1P is directly proportional to the partial molar S–V cross fluctuation density of 1P, SVδ1P, defined by eqn (2) and (3) in the Appendix. This signifies the effect of a solute on the mean square amplitude of the S–V cross fluctuation of bulk H2O.18,19 In other words, the mean square amplitude of the S–V cross fluctuation is monitored by perturbing the system by the infinitesimal increase of 1P. Thus, the behavior of HE1P1P, its increase/decrease, is directly proportional to that of SVδ1P. HEGlyGly, on the other hand, is only partially proportional to the equivalent SVδGly with an extra constant term.23 Unless the behavior of the latter constant term is known, that of HEGlyGly cannot be directly connected to SVδGly. Another practical disadvantage of the Gly-probe is that its point X is not as conspicuous as that of the 1P-probe, since the latter displays a peak top, while the former a bend point. (See Fig. 5 and 6 in the Appendix.) With these disadvantages we attempt to investigate how a hydrophile reacts to the subtle modification of H2O caused by the presence of ions within MS I, in comparison with a hydrophobe. Most of the solutes of biological significance are amphiphilic, and it would be important to investigate how hydrophobic and hydrophilic moieties respond to subtle modification caused by an ion while the basic integrity of liquid H2O is retained. As mentioned above, we have found that within the limited H2O-rich region the hydrophobic and hydrophilic moieties respond additively to H2O modification.16–23
Experimental
Glycerol (abbreviated as Gly in this paper) (Sigma, >99%) was degassed in vacuo at 80 °C for about 30 min and then charged into a 1000 μL syringe in a dry N2 atmosphere for the titration calorimetry described below. NH4Cl (Merck, >99.8%), N(CH3)4Cl (TMACl) (Merck, >98%) and Na(CH3COO) (NaOAc) (Sigma-Aldrich, >99.8%) were used to prepare stock solutions using Milli-Q water. The respective solutions were diluted to the desired initial mole fraction, x0S, immediately before use.The excess partial molar enthalpy of Gly, HEGly, is determined by using a TAM III isothermal titration calorimeter (TA Instruments, New Castle, DE, USA) at 25.000 ± 0.005 °C. The titration procedure was modified to enable facile delivery of highly viscous Gly as described in the previous work.24 Furthermore, a 30 min interval was given between successive titrations, in order to reduce a possible rheological effect of highly viscous Gly. The uncertainty in HEGly was estimated to be ±0.03 kJ mol−1.
Results and discussion
Fig. 1 shows the excess partial molar enthalpy of Gly, HEGly, in the ternary Gly–S–H2O at a given initial mole fraction of S in the mixed solvent, x0S. The raw data are given in Table S1 in the ESI.† While HEGly becomes more endothermic as x0S increases for TMACl, Fig. 1(b), and NaOAc, Fig. 1(c), that for NH4Cl shows a similar behavior at the low xGly range but becomes more exothermic at high xGly within the xGly range studied. But for all cases, the slopes of HEGly against xGly seem to become less as x0S increases. To see these trends more clearly, we evaluate HEGlyGly defined as,16,17,20–22| HEGlyGly ≡ N(∂HEGly/∂nGly) = (1 − xGly)(∂HEGly/∂xGly), | (1) |
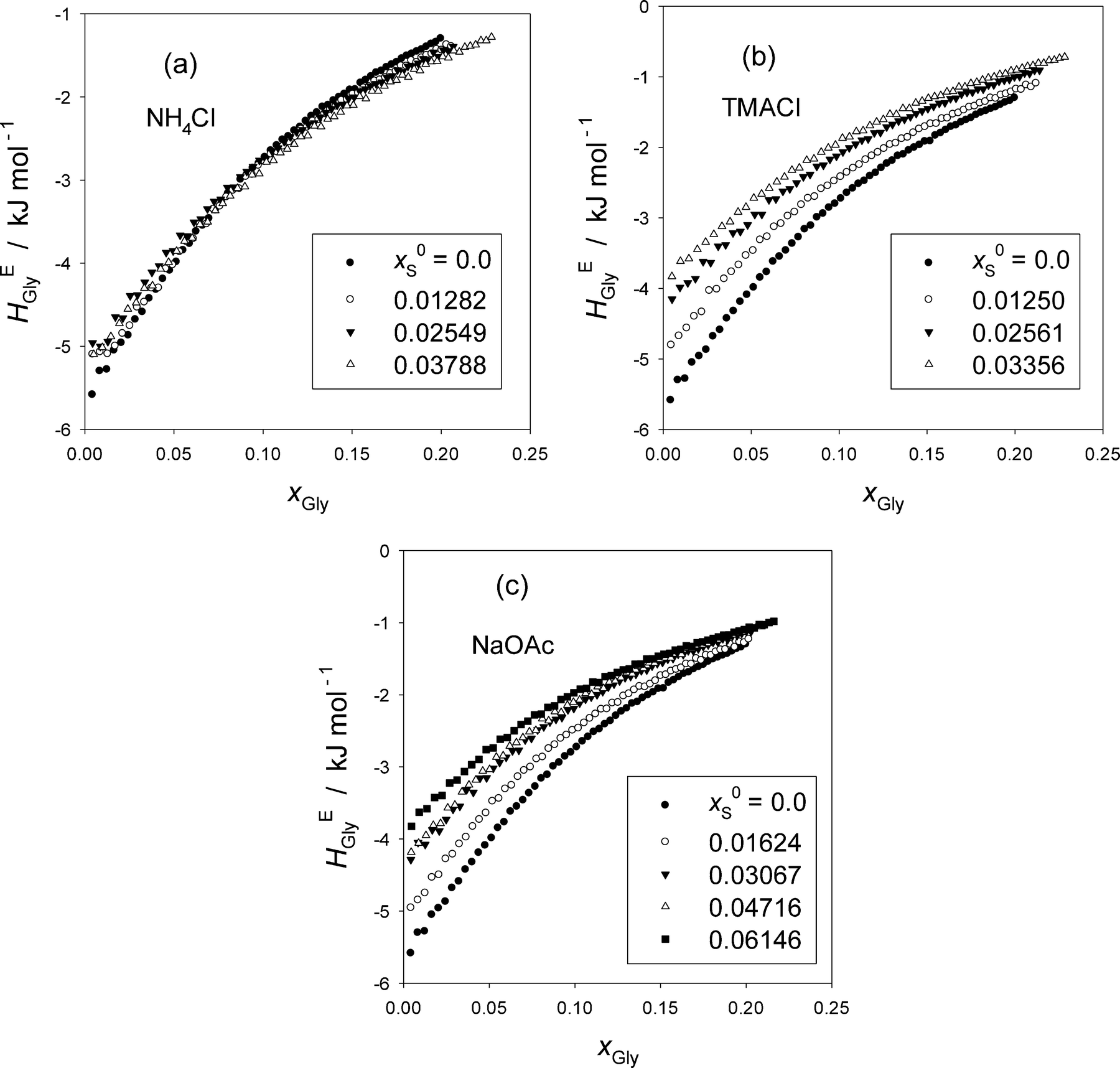 | ||
| Fig. 1 (a) Excess partial molar enthalpy of Gly, HEGly, in Gly–S–H2O (S = NH4Cl) at 25 °C. (b) Excess partial molar enthalpy of Gly, HEGly, in Gly–S–H2O (S = TMACl) at 25 °C. (c) Excess partial molar enthalpy of Gly, HEGly, in Gly–S–H2O (S = NaOAc) at 25 °C. | ||
The resulting HEGlyGly data are plotted in Fig. 2. Fig. 2(a) shows HEGlyGly for the binary Gly–H2O. It is apparent beyond the estimated uncertainty that the xGly-dependence pattern of HEGlyGly shows breaks in the slope at points X and Y at xGly = 0.073 and 0.14, respectively, indicated in the figure. The same behavior was observed in the previous Gly-probing study, though the xGly loci were at 0.08 and 0.015.24 The existence of the breaks at points X and Y was confirmed recently29 when we directly measured another third derivative quantity, the partial molar S–V cross fluctuation density of Gly in Gly–H2O, SVδGly, by differential pressure perturbation calorimetry.30 Since this third derivative quantity is determined directly, we could take one more derivative graphically. The resulting fourth derivative quantity showed the onset of a step anomaly correctly at xGly = 0.076 and its end at 0.14 at 25 °C.29 These should correspond to points X and Y in the third derivative quantity. As temperature increases, however, the step becomes progressively smaller and more obscure. The same observation was made in the previous Gly-probing study24 in that as S is added and x0S increases the break point X becomes more obscure to note in the HEGlyGly patterns.
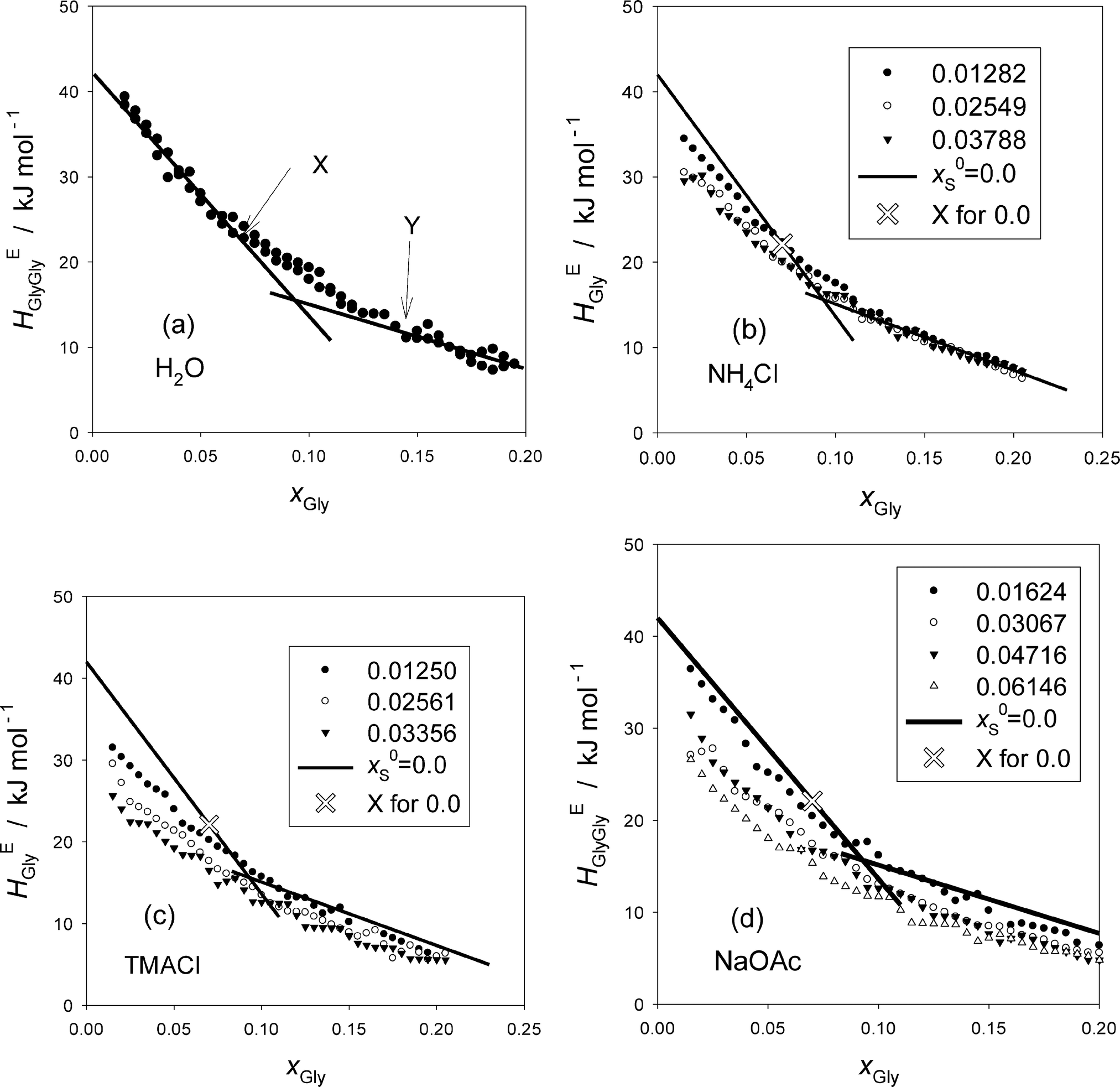 | ||
| Fig. 2 (a) Gly–Gly enthalpic interaction, HEGlyGly, in binary Gly–H2O at 25 °C. The value of point X for the binary Gly–H2O, x0Gly, was found to be 0.073 and that of Y 0.14. (b) Gly–Gly enthalpic interaction, HEGlyGly, in Gly–S–H2O at 25 °C for S = NH4Cl. (c) Gly–Gly enthalpic interaction, HEGlyGly, in Gly–S–H2O at 25 °C for S = TMACl. (d) Gly–Gly enthalpic interaction, HEGlyGly, in Gly–S–H2O at 25 °C for S = NaOAc. | ||
Fig. 2(b)–(d) show the results for the ternary Gly–S–H2O systems. The binary Gly–H2O system data are represented by two straight lines and its point X is indicated by a hollow X on the line. Point X is an important point that indicates the end of the dilute solution regime where the integrity of liquid H2O is lost. We found from our earlier studies20,23,31 that up to point X the integrity of liquid H2O is retained such that the hydrogen bond network is still connected throughout the bulk H2O. It is this dilute concentration range where the probing methodology by 1P or Gly is applicable.16,17 Thus, it is unfortunate that with the present data at hand the loci of point X are not located with confidence.
We thus approach differently. From the previous 1P-probing methodology, we found how each solute, a non-electrolyte or an individual ion, modifies H2O within the respective MS I. As mentioned above, Na+, NH4+ and Cl− belong to the class of “hydration centers” that are hydrated by 5.2, 1 and 2.3 molecules of H2O, respectively, but leave the bulk H2O away from hydration shells unperturbed. At least the same ions were shown not to alter the bulk H2O away from hydration shells by femto-second pump probe spectroscopic studies.5 OAc− is a “hydrophobe” that is hydrated by a total of 3.7 molecules of H2O. The hydrogen bond probability within the hydration shells is enhanced somewhat, but that of the bulk H2O away from the hydration shells is reduced progressively. The bulk H2O has not yet lost the hydrogen bond percolation until the system reaches point X.16,17,21
The distinction between hydration centers and hydrophobes was apparent in that the behavior of HE1P1P at x1P = 0 was different in the 1P-probing studies.16,17 Namely, for the hydration centers, the values of HE1P1P remain constant and independent of x0S, while they increased as x0S increased for the hydrophobes. Hence for salts consisting of counter ions in the “hydration center” such as, NaCl, and NH4Cl, they remained constant. For the present Gly-probing study, on the other hand, NH4Cl does not seem to stay constant as is evident in Fig. 2(b). The previous Gly-probing study24 indicates the same observation for NaCl also. This discrepancy between the 1P- and Gly-probing methodologies could be related to our findings that HE1P1P is directly proportional to the solute's effect on the S–V cross fluctuation density,16,17 while HEGlyGly is partially proportional with an extra constant term as discussed above and in the Appendix. The latter constant term, the origin of which is yet to be elucidated, may be responsible for the observed downward shift of HEGlyGly at xGly = 0 as x0S increases.
TMA+ was found to act as a hydrophile16,17,28 that forms hydrogen bonds directly with the existing (momentarily but perpetually) hydrogen bond network keeping the hydrogen bond connectivity intact. But it reduces the degree of fluctuation inherent in pure H2O progressively by breaking the proton donor–acceptor symmetry of liquid H2O. Probably reflecting this, Fig. 2(c) shows a little sharper decrease in the HEGlyGly value at xGly = 0, HEGlyGly (0), than the other two salts. NaOAc, on the other hand, with a hydrophobic OAc− ion shows a decrease in HEGlyGly (0), the value of HEGlyGly at xGly = 0. This contrasts with the behavior of HE1P1P observed in the 1P-probing study, where the equivalent HE1P1P (0) increased.16,17
To see these trends at xGly = 0 more clearly, the HEGlyGly data are extrapolated linearly to xGly = 0 and evaluated HEGlyGly (0) values. The results are plotted in Fig. 3(a). Also shown in the figure are the equivalent plots with hollow symbols taken from the previous Gly-probing study.24 For S = Na2SO4, the raw data were not used for the analysis in ref. 24. We reproduce the data here with the permission of the original authors as Table S2 in the ESI.† Both graphs of HEGly and HEGlyGly for S = Na2SO4 are also given as Fig. S3(a) and (b) (ESI†). Since the data points for HEGlyGly at xGly < 0.015 are not available for all cases, the uncertainty in the extrapolated results could amount to ±2 kJ mol−1. Fig. 3(b) shows the same plots for non-electrolyte samples. In the latter figure, two typical cases for hydrophobes, TBA and 1P, are shown. HEGlyGly (0) decreases as x0S increases, in contrast to the increase in HE1P1P (0), the value of HE1P1P at x1P = 0.16,17 This is only natural due to a geometrical reason. HE1P1P increases from x1P = 0 to point X, while HEGlyGly decreases down to its point X. A hydrophobic sample S will shift the HEBB pattern including point X towards west, a smaller value of xB (for B = 1P or Gly). Since a number, nH, of H2O molecules are used up for hydration, and they are not available for the probe B to interact, point X will be reached at a lesser value of xB. Indeed, the dynamics of the hydrating H2O was found to be several times slower than that of bulk H2O.5 This westward shift will inevitably result in an increase in the value of HEBB for a line with a positive slope (for B = 1P) and a decrease for that with a negative slope (for B = Gly), unless there is a mechanism to pin down HEBB (0) at a constant value. This is what happens for the 1P-probing, B = 1P, for “hydration centers”. Going back to Fig. 3(b), it is surprising that the decreases in HEGlyGly (0) for both TBA and 1P show no difference, although TBA is a stronger hydrophobe than 1P.16,17,20,21 This could indicate whether the Gly-probing is not as sensitive as 1P-probing or the effect of stronger TBA might be compensated for by its effect on the extra constant term discussed in the Appendix. Urea, a “hydrophile”, shows a marginal decrease in HEGlyGly (0) upon increasing its initial mole fraction, x0S. This could be understood by the fact that the hydrophilicity indices determined by the 1P-probing are similar for urea and the probe Gly; the values of b being −1210 and −1180 respectively.16
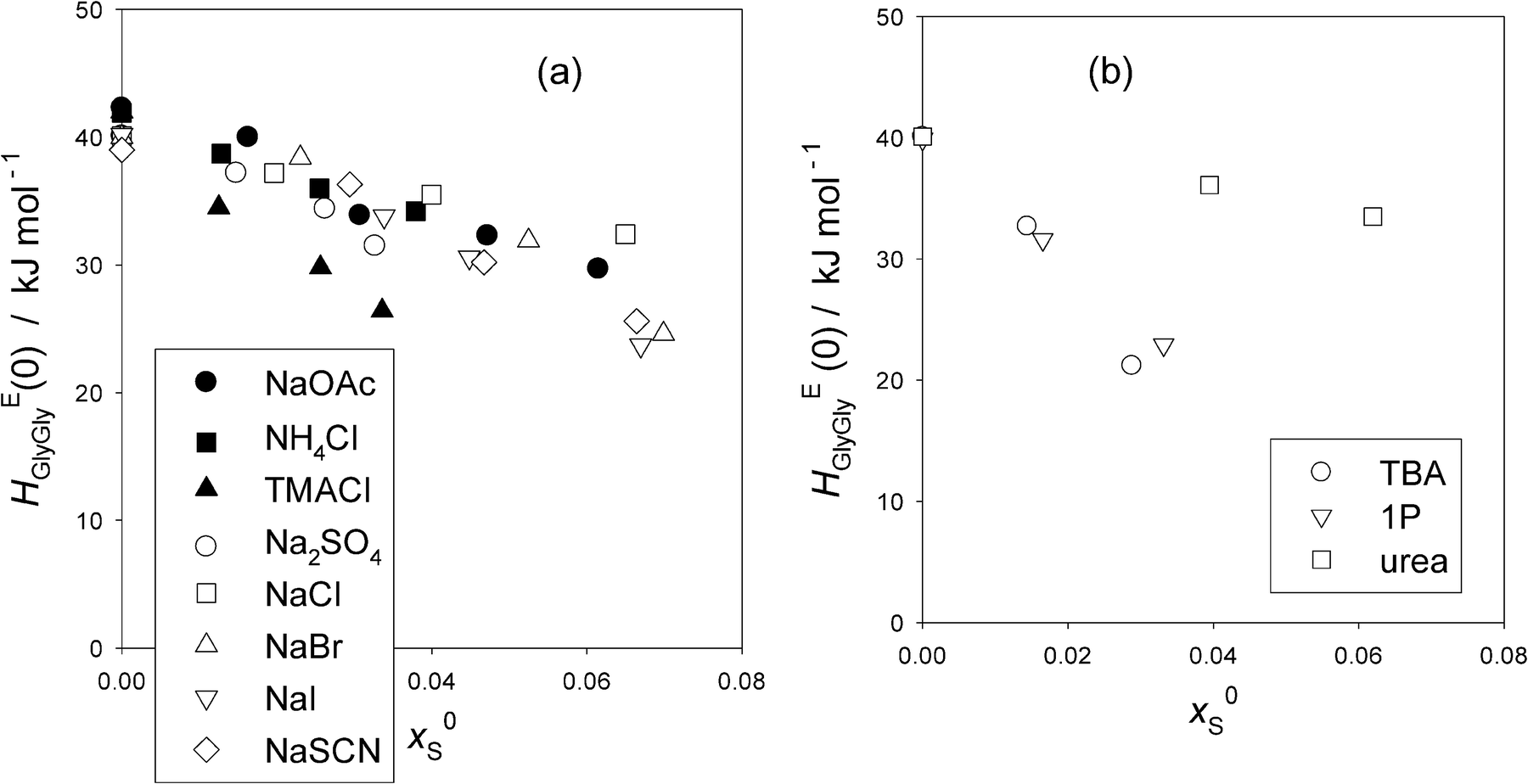 | ||
| Fig. 3 (a) The values of HEGlyGly at the infinite dilution, xGly = 0.0, HEGlyGly (0) against x0S for various salts (S). The uncertainty is estimated to be ±2 kJ mol−1. Filled symbols; this work, and hollow symbols; ref. 24. (b) The values of HEGlyGly at the infinite dilution, xGly = 0.0, HEGlyGly (0) against x0S for non-electrolytes. The data are taken from the previous Gly-probing study.24 The uncertainty is estimated to be ±2 kJ mol−1. | ||
Fig. 3(a) shows that for the hydration center salts, NH4Cl and NaCl, HEGlyGly (0) decreases slightly, by just above the uncertainty upon increasing x0S. They showed no change in HE1P1P (0) in the 1P-probing results.16,17 This decrease could also be due to an unknown effect on the extra constant term discussed above. Furthermore, there seems to be no difference among all these two hydration center salts in their x0S-dependence of HEGlyGly (0) in spite of the fact that the total hydration numbers are different; nH = 7.5 for NaCl and 3.3 for NH4Cl. This could also hint that the Gly-probe is not as sensitive as the 1P-probe towards subtle modification of H2O caused by the presence of S. NaOAc, containing a hydrophobic anion, shows no difference in the decrease of HEGlyGly (0) with those of hydration centers. Na2SO4, SO42− being a hydration center at x1P = 0 found by the 1P-probing,16,17 also shows the same trend. NaBr, NaI and NaSCN, consisting of Na+ and a hydrophilic anion with its hydrophilicity increasing in the order of Br− < I− < SCN−, do not show any difference among themselves nor from the hydration center group. TMACl is the only salt that stands out in terms of its decrease in HEGlyGly (0), as hydrophiles are expected to do. The hydrophilicity index of TMA+ is b = –1180, while those of other hydrophilic anions are −920, –2050, and –2800 respectively.16 Thus, TMA+ is only modestly hydrophilic, and yet the decrease of HEGlyGly (0) stands out. This must be due to the weaker effect of the counter ion Cl− than Na+. The hydration number, nH, for Cl− is 2.3, while that for Na+ is 5.2. But it is more likely that all these observations among salts could be due to the effect of each S on the extra constant term in the proportionality between HEGlyGly and the SVδGly.
Now that point X for the present HEGlyGly is hard to identify, we proceed our analysis by calculating the point X in the HEGlyGly pattern assuming that the shifts of xGly (X) and HEGlyGly (X) are both linear to x0S as was the case for the 1P-probing methodology.16,17 (X) indicates the respective coordinates at point X. Noting that the extrapolated value of x0S to xGly (X) = 0, x0S (0) is equal to 1/(nH + 1), and using the xGly locus of point X for the binary Gly–H2O determined in Fig. 2(a), we calculated the xGly-loci of point X at given x0S, which are listed in Table 1 for the present data. The same data treatment is applied to the previous Gly-probing study,24 and listed in Table S4 in the ESI.† We then read off the value of HEGlyGly in Fig. 2(b)–(d) for the present data and equivalent graphs of HEGlyGly against xGly for the previous work24 at the calculated point X, xGly (X). The HEGlyGly (X) values are also listed in Table 1 and Table S4 (ESI†), and plotted in Fig. 4(a) for salts, and in Fig. 4(b) for non-electrolytes. The uncertainty of the resulting HEGlyGly (X) is estimated to be ±2 kJ mol−1.
| Salt | n H | x 0S | x Gly (X) | H EGlyGly (X) | |
|---|---|---|---|---|---|
| NH4Cl | + | 1 | 0 | 0.073 | 21.1 |
| − | 2.3 | 0.01282 | 0.06898 | 21 | |
| (tot) | 3.3 | 0.02549 | 0.06500 | 21 | |
| 0.03788 | 0.06111 | 21 | |||
| TMACl | + | 0 | 0 | 0.073 | 21.1 |
| − | 2.3 | 0.01250 | 0.06999 | 20 | |
| (tot) | 2.3 | 0.02561 | 0.06683 | 18 | |
| 0.03356 | 0.06492 | 17 | |||
| NaOAc | + | 5.2 | 0 | 0.073 | 21.1 |
| − | 3.7 | 0.01624 | 0.06126 | 23 | |
| (tot) | 8.9 | 0.03067 | 0.05084 | 22 | |
| 0.04716 | 0.03892 | 23 | |||
| 0.06146 | 0.02852 | 23 | |||
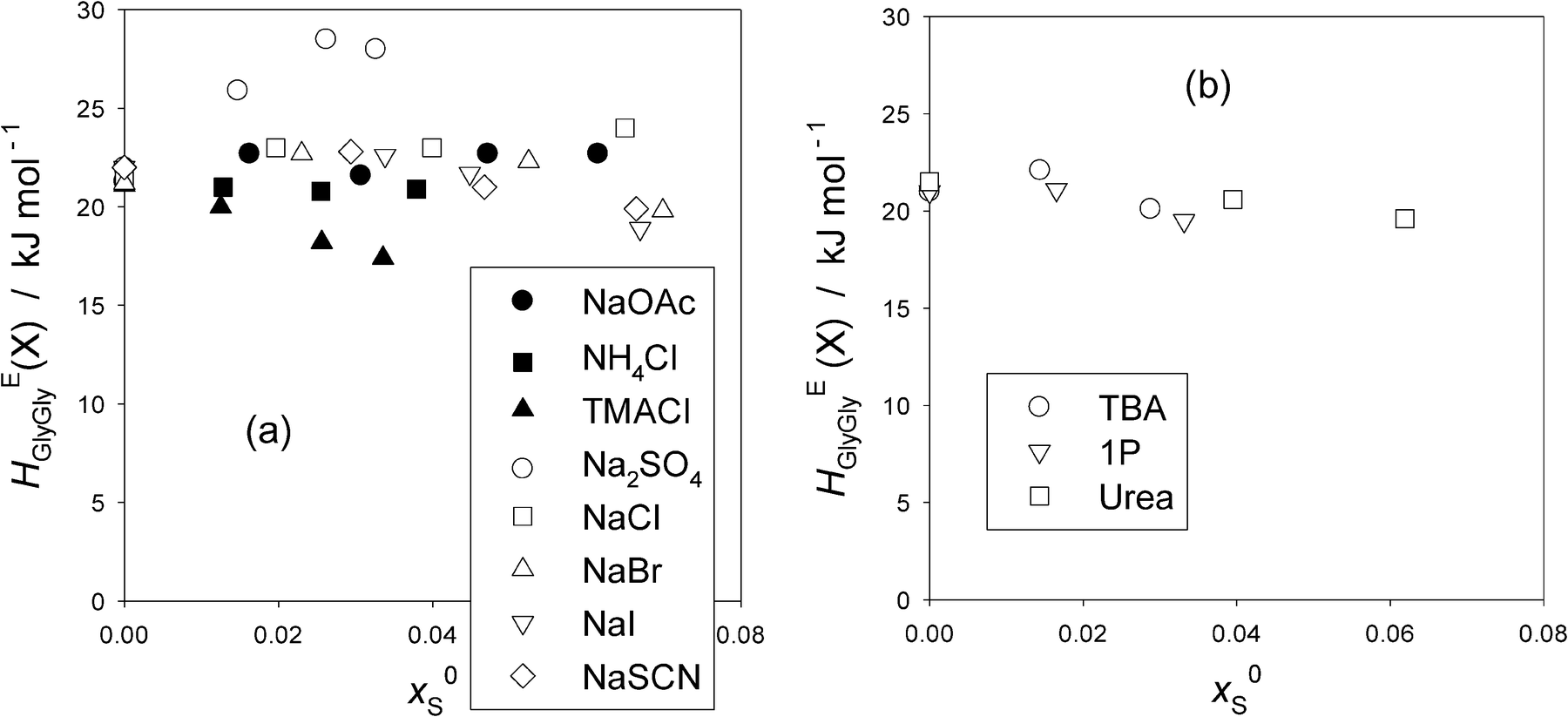 | ||
| Fig. 4 (a) The values of HEGlyGly at presumed point “X”, HEGlyGly (X) against x0Sfor various salts (S). Filled symbols; present work and hollow symbols; ref. 24. The uncertainty is estimated to be ±2 kJ mol−1. (b) The values of HEGlyGly at presumed point “X”, HEGlyGly (X), against x0S for non-electrolytes. Evaluated using the data in the previous Gly-probing study.24 The uncertainty is estimated to be ±2 kJ mol−1. | ||
For all other salts in Fig. 4(a) except for Na2SO4 and TMACl, HEGlyGly (X) may be regarded as remaining constant and independent of x0S, taking into account the estimated uncertainty, ±2 kJ mol−1. For Na2SO4, HEGlyGly (X) clearly increases as x0S increases. This is an interesting and important finding. From the 1P-probing, SO42− was found to belong to a special case of the “hydration center”.16,32 As the mole fraction of the probe 1P, x1P, increases, both S and 1P together were found to reduce the hydrogen bond probability of bulk H2O just as a hydrophobe stronger than the probe 1P does, while in the absence of 1P (i.e. at x1P = 0), SO42− alone acts as purely a hydration center. Thus the present finding suggests that the increase in HEBB at point X is independent of the identity of the probe B. Namely, as xB increases and hence the available bulk H2O decreases, there must be some inherent mechanisms due only to SO42− to reduce the hydrogen bond probability of bulk H2O. Self-aggregation of SO42− could be a reason, as observed for urea above x0S > 0.08 (S = urea),33 for the 1-butyl-2,3-dimethylimidazolium cation at x0S > 0.00634 and for the 1-butyl-3-methylimidazolium cation at x0S > 0.014.35 However, they all show a sudden decrease in the slope of HEBB (X) vs. x0S in the respective 1P-probing studies. For Na2SO4 aqueous solutions, a dielectric relaxation study suggests the formation of H2O separated cation–anion pairing as its concentration increases,36 but this would also reduce HEBB (X) rather than increase as observed here. We rather speculate that as the availability of un-hydrated bulk H2O decreases, SO42− ions may start to interact more strongly with the existing hydrogen bond network of bulk H2O rather than just forming hydration shells. This may be due to the fact that SO42− ions presumably spread O atoms out in four tetragonal directions. As a result, the average hydrogen bond probability of bulk H2O is reduced progressively. Recent studies using modern non-linear spectroscopic techniques aided by MD simulations37,38 revealed how the ClO4− ion exchanges hydrogen bonds from a H2O molecule with another in concentrated aqueous solutions of about 0.1 mole fraction. At this concentration, there are therefore hardly any H2O molecules left to study the state of bulk H2O away from hydration shells. Similar studies on SO42− in H2O could provide an important clue with more dilute aqueous solutions so that the state of bulk H2O away from hydration shells could be studied.
In the case of TMACl, TMA+ being hydrophilic, slightly more so than Gly16,28 and Cl− being a weak hydration center,16,17 the decrease in HEGlyGly (X) reflects the effect of TMA+ and indicates the reduction in the effect of the solute on the degree of fluctuation in the bulk H2O.
For NH4Cl and NaCl, the constituent ions are all hydration centers. Hence, these salts do not alter the bulk H2O away from hydration shells, and hence the effect of the solute on the degree of the S–V cross fluctuation should remain constant independent of x0S. This is exactly what we observe in Fig. 4(a). For the 1P-probe, however, not only at point X but also at xB = 0 the values of HEBB were found to remain constant. For the present Gly-probe, the values of HEGlyGly (0) at xGly = 0 do not remain constant, as shown in Fig. 3(a). OAc−, on the other hand, was found to act as a hydrophobe with the total nH = 3.7 and to reduce the hydrogen bond probability of bulk H2O to the same degree as the probe 1P.16,17,27Fig. 4(b) indicates the behavior of typical hydrophobes, TBA and 1P. They are hydrated by 20 and 29 H2O molecules, respectively,16 and reduce the hydrogen bond probability of bulk H2O away from hydration shells, more so for TBA than 1P. As a consequence, the effect of the solute on the degree of S–V cross fluctuation density increases due to a decrease in the negative contribution. Namely, as the hydrogen bond probability of liquid H2O decreases, the chances for local and instantaneous formation of highly hydrogen bonded patches which contributes negatively to the S–V cross fluctuation decrease. Thus the net fluctuation increases, which should manifest in an increase in HEBB. It was indeed the case for the 1P-probing, B = 1P, and the value of HEBB (X) is larger for TBA at point X than for 1P.16Fig. 4(b) shows, on the other hand, that for the Gly-probing, the values of HEGlyGly (X) remain constant, independent of x0S for both hydrophobes. Similarly, the values of HEGlyGly (X) for NaOAc remain constant as observed in Fig. 4(a).
The remaining three Na-salts are made of hydrophilic anions as found by the 1P-probing.16,17 The hydrophilicity is stronger in the order of SCN− > I− > Br−, the values of hydrophilicity being b = −2800, −2050, and −920 respectively.16 In spite of the almost three-fold difference, the distinction in the x0S-dependence of HEGlyGly (X) among them is not apparent. Thus, the Gly-probe appears to be insensitive to the difference in the modified H2O by hydrophiles as well as by hydrophobes. Or it could be due to the constant additive term in the partial proportionality of the partial molar S–V cross fluctuation and HEGlyGly that makes the HEGlyGly (X) appear insensitive.
Thus, while the extra constant term in the partial proportionality between HEGlyGly and SVδGly must be measured and its nature ought to be elucidated, we suggest that the behavior of HEGlyGly is not entirely inconsistent with the effects of S on H2O deduced by the 1P-probing methodology.16,17 It is clear, however, that the Gly-probe is not so sensitive as the 1P-counterpart. This would have an important implication for understanding the Hofmeister rankings, in that it is the hydrophobic part of a biological polymer that will respond more strongly to the slight modification of liquid H2O caused by the presence of an ion.
Appendix
Fig. 5 shows the plots of HE1P1P and SVδ1P for the binary 1P–H2O system. The ordinate for SVδ1P is scaled by a single factor ξ. The definition of HE1P1P is given in eqn (1) in the main text except for swapping subscripts Gly with 1P. HE1P1P signifies the 1P–1P interaction in terms of enthalpy in the solution. The S–V cross fluctuation density, SVδ, is defined as,18,19,23| SVδ ≡ 〈(ΔS)(ΔV)〉/k〈V〉 = Tαp. | (2) |
| SVδ1P ≡ N(∂SVδ/∂n1P) = (1 − x1P)(∂SVδ/∂x1P). | (3) |
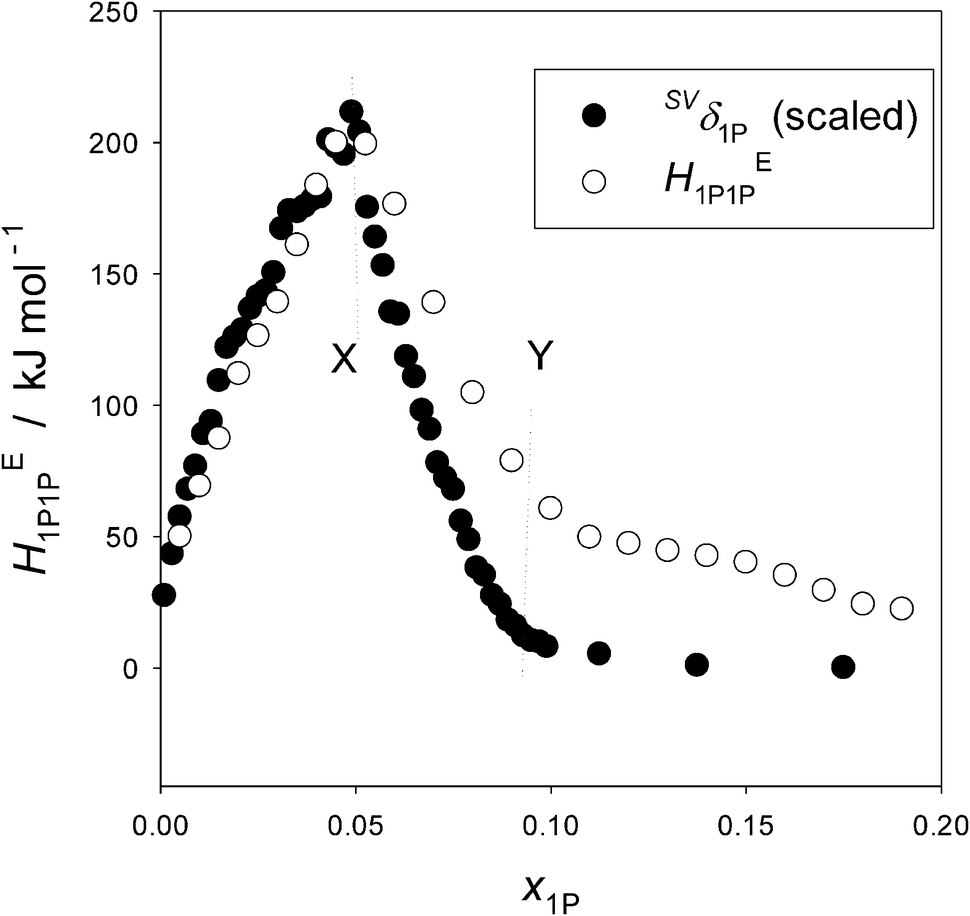 | ||
| Fig. 5 The partial molar S–V cross fluctuation density of 1P, SVδ1P, and the enthalpic 1P–1P interaction, HE1P1P, in 1P–H2O at 25 °C. The ordinate for SVδ1P is scaled by a single factor. Reproduced with permission from ref. 18. Copyright (1999), NRC Research Press, National Research Council of Canada, Ottawa. | ||
What Fig. 5 indicates then is that HE1P1P and SVδ1P are directly proportional with a single factor ξ within the dilute region up to point X; namely,
| HE1P1P = ξSVδ1P. | (4) |
The equivalent quantities of binary Gly–H2O, HEGlyGly and SVδGly are plotted in Fig. 6. The raw data of αp determined by dilatometry40 are used to calculate SVδ. A clear distinction is evident between Fig. 5 and 6 in their xB-dependence patterns. The former pattern is unique to a hydrophobic solute, 1P, and the latter to a hydrophilic solute, Gly. Or rather, we used this qualitative difference in the xB-dependence pattern of HEBB to distinguish “hydrophobes” and “hydrophiles”. Furthermore, HEGlyGly is only partially proportional to SVδGly up to point X. Namely,
| HEGlyGly = ηSVδGly + ζ, | (5) |
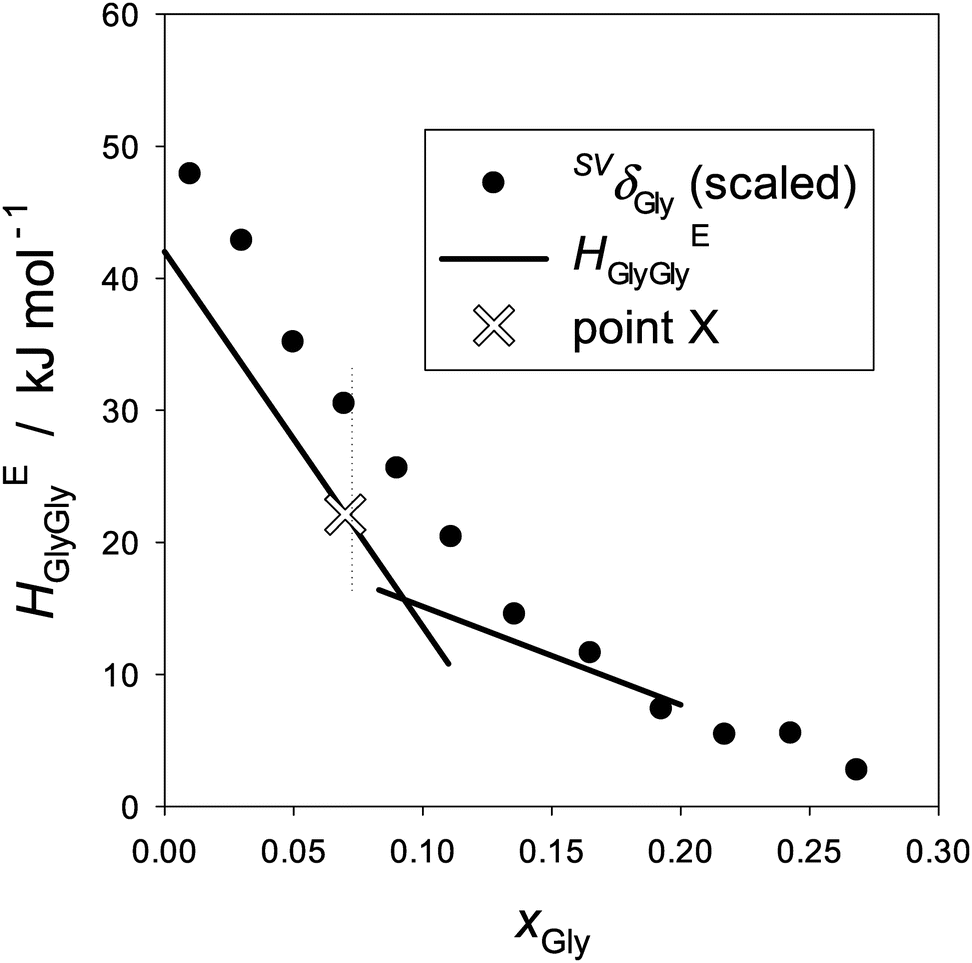 | ||
| Fig. 6 The partial molar S–V cross fluctuation density of Gly, SVδGly, and the Gly–Gly enthalpic interaction, HEGlyGly, in binary Gly–H2O at 25 °C. The ordinate for SVδGly is scaled by a single factor. The data of αp were taken from ref. 41. HEGlyGly data are represented by two straight lines as shown in Fig. 2(b)etc. | ||
Acknowledgements
This work was supported by the Carlsberg Foundation.References
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1887, XXIV, 247–260 Search PubMed.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, XXV, 1–30 Search PubMed.
- W. Kunz, J. Henle and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 19–37 CrossRef CAS PubMed.
- K. D. Collins and M. W. Washabaugh, Q. Rev. Biophys., 1985, 18, 323–422 CrossRef CAS.
- H. L. Bakker, Chem. Rev., 2008, 108, 1456–1473 CrossRef CAS PubMed.
- I. A. Heisler, K. Mazur and S. R. Meech, J. Phys. Chem. B, 2011, 115, 1863–1973 CrossRef CAS PubMed.
- I. A. Heisler and S. R. Meech, Science, 2010, 327, 857–860 CrossRef CAS PubMed.
- I. Waluyo, C. Huang, D. Nordlund, U. Bergmann, T. M. Weiss, L. G. M. Petersson and A. Nilsson, J. Chem. Soc., 2010, 134, 064513 Search PubMed.
- C. D. Cappa, J. D. Smith, K. R. Wilson, B. M. Messer, M. K. Gilles, R. C. Cohen and R. J. Saykally, J. Phys. Chem. B, 2005, 109, 7046–7052 CrossRef CAS PubMed.
- Y. S. Lin, B. M. Auer and J. L. Skinner, J. Chem. Phys., 2009, 131, 144511 CrossRef PubMed.
- J. D. Smith and R. L. Saykally, J. Am. Chem. Soc., 2007, 129, 13847–13856 CrossRef CAS PubMed.
- J. Paterová, K. B. Rembert, J. Heyda, Y. Kurra, H. I. Okur, W. R. Liu, C. Hilty, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2013, 117, 8150–8158 CrossRef PubMed.
- A. Salis, F. Cugia, D. F. Parsons, B. W. Ninham and M. Monduzzi, Phys. Chem. Chem. Phys., 2012, 14, 4343–4346 RSC.
- Y. Zhamg and P. S. Cremer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15249–15253 CrossRef PubMed.
- P. Lo Nostro and B. W. Ninham, Chem. Rev., 2012, 112, 2286–2322 CrossRef CAS PubMed.
- Y. Koga, Phys. Chem. Chem. Phys., 2013, 15, 14548–14565 RSC.
- Y. Koga, Solution Thermodynamics and Its Application to Aqueous Solutions: A Differential Approach, Elsevier B.V., Amsterdam, 2007, ch. VII and VIII, pp. 175–239 Search PubMed.
- Y. Koga, Can. J. Chem., 1999, 77, 2039–2045 CrossRef CAS.
- Y. Koga, Solution Thermodynamics and Its Application to Aqueous Solutions: A Differential Approach, Elsevier B.V., Amsterdam, 2007, Section V-3, pp. 117–130 Search PubMed.
- Y. Koga, J. Phys. Chem., 1996, 100, 5172–5181 CrossRef CAS.
- Y. Koga, Solution Thermodynamics and Its Application to Aqueous Solutions: A Differential Approach, 2007, ch. V, pp. 89–150 Search PubMed.
- Y. Koga, Solution Thermodynamics and Its Application to Aqueous Solutions: A Differential Approach, Elsevier B.V., Amsterdam, 2007, ch. VI, pp. 151–173 Search PubMed.
- Y. Koga, Solution Thermodynamics and Its Application to Aqueous Solutions: A Differential Approach, Elsevier B.V., Amsterdam, 2007, Section VI-3, pp. 160–166 Search PubMed.
- P. Westh, E. L. Rasmussen and Y. Koga, J. Solution Chem., 2011, 40, 93–105 CrossRef CAS.
- Y. Koga, K. Nishikawa and P. Westh, J. Phys. Chem. A, 2004, 108, 3873–3877 CrossRef CAS.
- Y. Koga, Solution Thermodynamics and Its Application to Aqueous Solutions: A Differential Approach, Elsevier B.V., Amsterdam, 2007, Section IV-4, pp. 78–84 Search PubMed.
- T. Kondo, Y. Miyazaki, A. Inaba and Y. Koga, J. Phys. Chem. B, 2012, 116, 3571–3577 CrossRef CAS PubMed.
- Y. Koga, F. Sebe and K. Nishikawa, J. Phys. Chem. B, 2013, 117, 877–883 CrossRef CAS PubMed.
- K. Yoshida, A. Inaba and Y. Koga, J. Solution Chem. Search PubMed , accepted.
- K. Yoshida, S. Baluja, A. Inaba, K. Tozaki and Y. Koga, J. Solution Chem., 2011, 40, 1271–1278 CrossRef CAS.
- Y. Koga, Netsu Sokutei, 2003, 30, 54–65 CAS . Available in a pdf file on request to the author: E-mail: koga@chem.ubc.ca.
- Y. Koga, T. Kondo, Y. Miyazaki and A. Inaba, J. Solution Chem., 2012, 41, 1388–1400 CrossRef CAS.
- Y. Koga, Y. Miyazaki, Y. Nagano and A. Inaba, J. Phys. Chem. B, 2008, 112, 11341–11346 CrossRef CAS PubMed.
- H. Kato, K. Miki, T. Mukai and K. Nishikawa, J. Phys. Chem. B, 2009, 113, 14754–14760 CrossRef CAS PubMed.
- K. Miki, P. Westh, K. Nishikawa and Y. Koga, J. Phys. Chem. B, 2005, 109, 9014–9019 CrossRef CAS PubMed.
- R. Buchner, S. G. Capewell, G. Hefter and P. M. May, J. Phys. Chem. B, 1999, 103, 1185–1192 CrossRef CAS.
- M. Ji, M. Odelius and K. J. Gaffney, Science, 2010, 328, 1003–1005 CrossRef CAS PubMed.
- S. Park, M. Odelius and K. J. Gaffney, J. Phys. Chem. B, 2009, 113, 7825–7835 CrossRef CAS PubMed.
- Y. Koga, J. Chem. Phys., 2012, 137, 124503 CrossRef PubMed.
- E. C. H. To, J. V Davies, M. Tucker, P. Westh, C. Trandum, K. S. H. Suh and Y. Koga, J. Solution Chem., 1999, 28, 1137–1157 CrossRef CAS.
- J. Lyklema, Chem. Phys. Lett., 2009, 467, 217–222 CrossRef CAS PubMed.
- Y. Levin, A. P. dos Santos and A. Diehl, Phys. Rev. Lett., 2009, 103, 257802 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53474j |
| This journal is © the Owner Societies 2014 |