Catalytic alkene cyclization reactions for the stereoselective synthesis of complex “terpenoid-like” heterocycles†
Jared T.
Moore
,
Cristian
Soldi
,
James C.
Fettinger
and
Jared T.
Shaw
*
Department of Chemistry, University of California, 1 Shields Ave, Davis, CA 95618, USA. E-mail: jtshaw@ucdavis.edu; Tel: +1-530-752-9979
First published on 25th October 2012
Abstract
A new catalytic synthesis of densely substituted tetrahydroquinolines is described. This reaction forms up to two rings, three bonds, and three stereogenic centers with excellent stereo- and regiocontrol in a single step. Although control experiments demonstrate that the active catalyst is protic acid, Sc(OTf)3 serves as an effective and practical pre-catalyst. The scope of this reaction is demonstrated with 21 monocyclizations and 14 bicyclization reactions.
Introduction
Cyclization reactions of carbocations are powerful methods for the synthesis of fused polycyclic organic molecules.1 After the proposed biosynthesis of steroids was elucidated,2 Johnson employed polycyclization reactions in the synthesis of steroids and related carbocyclic structures.3 The strategy involves an initiating electrophile that produces a cationic center and terminating nucleophile, usually an alkene, alkyne or an electron-rich aromatic ring.Subsequent initiators have included epoxides,4 halonium ions,5 episulfonium ions,6 tertiary carbocations (from alkene protonation7 or addition to an oxonium ion8), and vinyl cations formed by the mercuration9 or auration10 of alkynes. Similar frameworks can also be assembled via radical cascade reactions.11 In spite of the wide use of this type of reaction to prepare polycyclic hydrocarbons, catalytic alkene cyclization reactions that produce heterocycles are less common than their all-carbon counterparts.7c,10,12–16 To date, there is a single example of a polycyclization that forms a complex nitrogen-based heterocycle from a halonium-ion induced reaction.17 Herein we report broadly applicable mono- and bicyclization reactions that form densely substituted tetrahydroquinolines and related tricyclic analogs of diterpenoids (Fig. 1).
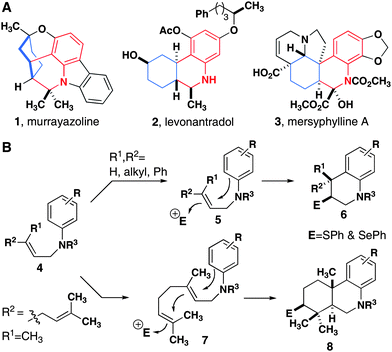 | ||
| Fig. 1 (A) Densely substituted tetrahydroquinolines in the core structures of natural products (1 and 3) and a synthetic analog of THC (2). (B) Electrophile-induced mono- and bicyclization reactions. | ||
As part of our continuing interest in the exploration and modification of natural products that mediate biological processes, we sought to develop a cyclization reaction that produces nitrogen-containing heterocyclic analogs of terpenoids with high diastereoselectivity and in high yield. In addition to the assembly of complex core structures, we were specifically interested in reactions that would enable the products to be used in subsequent reactions on the ring where the cyclization is initiated. With this goal in mind, we chose N-geranyl anilines appended with various protecting groups as our initial substrates and examined reactions that installed heteroatoms.
Results and discussion
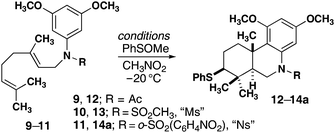
Cyclizations using an electrophilic sulfur reagent (PhSOCH3) developed by Livinghouse were well-suited to our substrates. Initial studies focused on the use of PhSOCH3 in the presence of a Lewis acid. Although, efficient bicyclization was initially observed with four equivalents of BF3·OEt2, a single equivalent of Sc(OTf)3 was found to be superior (Table 1, entry 1).18 Tricyclic product 12 was formed in high yield as a single diastereomer. X-ray crystallographic analysis of a crystalline derivative revealed that the stereochemistry of the product is in direct analogy to carbocyclization reactions that proceed through “chair–chair” conformations and lead to trans-fused products. Although the cyclization of 9 was optimized to proceed with a single equivalent of Sc(OTf)3, catalysis was not observed with this Lewis acid or any others examined. Replacement of acetyl (Ac) with the less basic methanesulfonyl (Ms) group enabled the reaction to proceed in the presence of catalytic quantities of Sc(OTf)3 (Table 1, entries 5 and 6). A control experiment with DTBP suggests that the true catalyst is protic acid produced by adventitious moisture (Table 1, entry 7). A scan of protic acids revealed the necessity of strong acids such as TfOH. Given the practical advantage of handling Sc(OTf)3, we settled on this reagent as the catalyst of choice.19,20 Finally, replacement of Ms with Ns (11, Table 1 entry 11) enabled a high yield of cyclization product with the added flexibility of facile cleavage of this group. Related reactions with epoxygeranyl starting materials failed to produce cyclization products.|
|
|||
|---|---|---|---|
| Entry | Substrate (R) | Conditionsa | Yield |
| a Substrate concentration at 0.1 M unless otherwise noted. b Yield calculated by HPLC with an internal standard curve. c DTBP (di-tert-butyl-4-methylpyridine) was added as a Brønsted acid scavenger. | |||
| 1 | 9 (Ac) | 1.0 equiv. Sc(OTf)3 | 90% |
| 2 | 9 (Ac) | 0.5 equiv. Sc(OTf)3 | 44% |
| 3 | 10 (Ms) | 1.0 equiv. Sc(OTf)3 | 0%b |
| 4 | 10 (Ms) | 0.3 equiv. Sc(OTf)3 | 32%b |
| 5 | 10 (Ms) | 0.1 equiv. Sc(OTf)3 | 52%b |
| 6 | 10 (Ms) | 0.1 equiv. Sc(OTf)3 (0.02 M 10) | 86%b |
| 7 | 10 (Ms) | 0.1 equiv. Sc(OTf)3 | 0%b |
| 0.1 equiv. DTBP, (0.02 M 10)c | |||
| 8 | 10 (Ms) | 0.05 equiv. TfOH (0.02 M 10) | 57%b |
| 9 | 10 (Ms) | 0.2 equiv. pTSA (0.02 M 10) | 18%b |
| 10 | 10 (Ms) | 0.1 equiv. Tf2NH (0.02 M 10) | 44%b |
| 11 | 11 (Ns) | 0.1 equiv. Sc(OTf)3 (0.02 M 11) | 89% |
Monocyclization reactions also proceed in high yield. Initial experiments using the optimized conditions for 11 with substrate 15 (Scheme 1) produced acyclic compound 17 resulting from addition of methoxide to the episulfonium ion. Although the analogous acyclic product 16 was also formed with PhSCl, a significant quantity of the desired cyclization product 18a was also observed. Changing from dichloromethane to more polar solvents resulted in complete cyclization to form 18a in 73% and 87% yields from acetonitrile and nitromethane, respectively. High reactivity was also observed with phenylselenium chloride (PhSeCl), producing 18b from 15 in 78% yield in dichloromethane. The yield of cyclized product 18b was 83% with the addition of NaSbF6 as a chloride scavenger and was increased to 95% by switching to nitromethane as solvent.
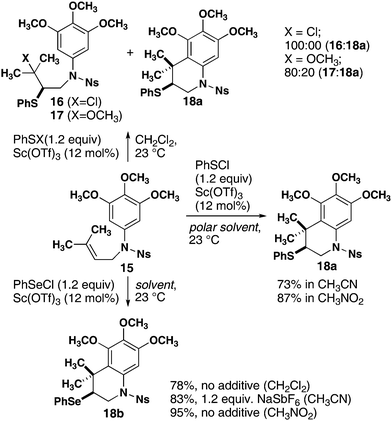 | ||
| Scheme 1 Optimization of monocyclization reactions mediated by sulfur and selenium electrophiles. | ||
Optimized cyclization conditions were effective for a variety of prenylated aniline substrates (Table 2). Electron-rich anilines, i.e. those with at least one alkoxy group in the para position to the reacting carbon center were cyclized in 68–88% yield. In the case of 29, 30, and 32 in which two possible regioisomers could be formed, only one detectable isomer was observed. Although an interaction between the fused cyclohexyl ring of 27 reduced the efficiency of the formation of 36, the meta methyl groups of 25 did not negatively impact the formation of 34, in which a quaternary carbon is added to the ortho position. Substitution with a single meta methyl group (26) resulted in efficient cyclization with modest regioselectivity. The origin of higher regioselectivity resulting from a more electron rich aromatic ring is not immediately obvious, given that the reactions were conducted at similar temperatures.
| a Solvent was acetonitrile with 1.2 equivalent of NaSbF6 added. b Yield over two steps after removal of Ns group with K2CO3/PhSH. |
|---|
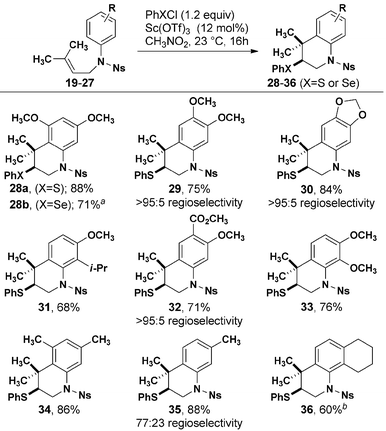
|
We next investigated the monocyclization reactions using various substituted alkenes (Table 3). Allyl substituted sulfonamide 37 was inert to cyclization with PhSCl and cyclized in good yield with PhSeCl. N-Cinnamyl substrate 38 reacted smoothly to provide a single detectable diastereomer of 42 and the expected anti-configuration was confirmed by X-ray crystallography. Alkyl-substituted internal alkenes E- and Z-39 exhibited lower overall reactivity when compared to phenyl-substituted 38. Cyclization of E-39 gave exclusively anti-products with both PhSCl and PhSeCl. The analogous reaction of Z-39 produced a mixture when treated with PhSeCl, suggesting that the intermediate selenonium ion undergoes isomerization faster than cyclization. The same cyclization mediated by PhSCl proceeded with high diastereoselectivity to give syn-44a. Finally, installation of an allylic stereogenic center completely controlled the formation of the two new stereogenic centers formed on cyclization of 40 to 45.
| a Temperature was −20 °C. |
|---|
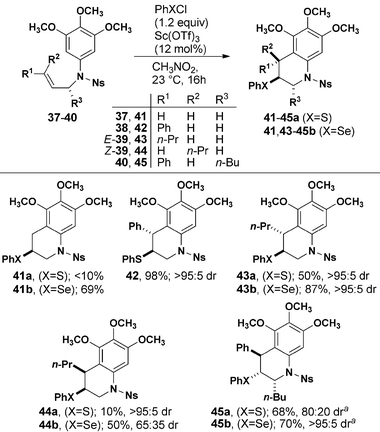
|
The cyclization reaction exclusively forms 6-membered rings. Methallyl substrate 46, which can conceivably form either 5- or 6-membered ring products, was subjected to the standard conditions (Scheme 2). Only starting material and small amounts of seleno-halogenation of the double bond were observed. In contrast, homoallylic substrate 47 resulted in exo-cyclization and produced tetrahydroquinoline 48 in high yield.
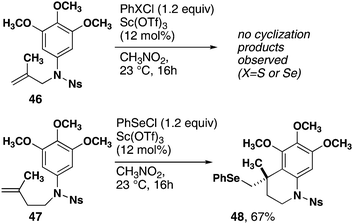 | ||
| Scheme 2 Monocyclization reactions of methallyl- and homomethallyl-substrates. | ||
Optimized conditions for the monocyclization were employed in the bicyclization of geranylated aniline sulfonamides (Table 4). Overall, narrower substrate scope was observed and in nearly every case. PhSeCl as a mediator resulted in higher yields than PhSCl. Electron-rich substrates 49–51, produced tricyclic compounds 57–59 in excellent yields. The less activated 52–54 were cyclized in 32–51% yield. In several cases (60–63), purification was complicated by co-elution of starting material and product, which was avoided by purifying after removing the nitrobenzenesulfonyl (Ns) group. A notable difference between the mono- and bicyclization reactions is the lack of regioselectivity in the latter relative to the former. Substrates 55 and 56 produce 63 and 64, respectively, with little preference for attack at the ortho or para positions relative to the alkoxy groups.
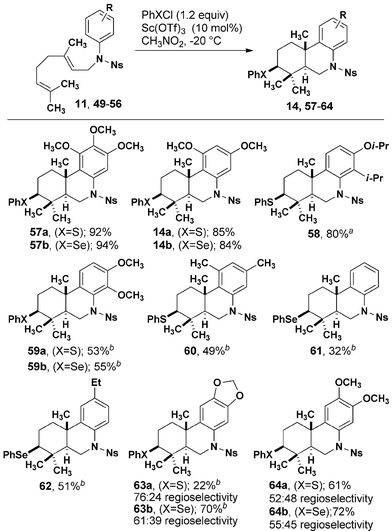
Conclusions
In conclusion, we have developed the first catalytic alkene cyclization reaction mediated by sulfur- and selenium-based electrophiles that produces heterocycles. Monocyclization reactions proceed with high levels of diastereo- and regioselectivity. Excellent substrate control was observed when a substrate with an allylic stereogenic center was employed. Although bicyclization reactions also proceeded in high yield in many cases, the regioselectivity for attack of the aromatic ring was lower than that observed for monocyclization. This new method provides a superb platform for the synthesis of densely substituted tetrahydroquinolines that should prove useful for the synthesis of natural products and nitrogen analogs of diterpenoids.Acknowledgements
This work was supported by the NIH/NIAID (R01AI08093) and by NIH/NIGMS (T32-GM008799), which provided JTM with predoctoral fellowship support. JTM was also supported by a Bradford Borge fellowship from UC Davis. CS thanks CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) and the Brazilian Ministry of Education for a postdoctoral fellowship.Notes and references
- Polycyclization reactions were recently reviewed: R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730–4756 CrossRef CAS.
- (a) R. B. Woodward and K. Bloch, J. Am. Chem. Soc., 1953, 75, 2023–2024 CrossRef CAS; (b) P. A. Stadler, A. Eschenmoser, H. Schinz and G. Stork, Helv. Chim. Acta, 1957, 40, 2191–2198 CrossRef CAS.
- W. S. Johnson, S. Escher and B. W. Metcalf, J. Am. Chem. Soc., 1976, 98, 1039–1041 CrossRef CAS.
- E. J. Corey, G. Luo and L. S. Lin, J. Am. Chem. Soc., 1997, 119, 9927–9928 CrossRef CAS.
- (a) A. Sakakura, A. Ukai and K. Ishihara, Nature, 2007, 445, 900–903 CrossRef CAS; (b) M. Uyanik, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 5649–5652 CrossRef CAS.
- (a) E. Edstrom and T. Livinghouse, J. Am. Chem. Soc., 1986, 108, 1334–1336 CrossRef CAS; (b) S. R. Harring and T. Livinghouse, J. Org. Chem., 1997, 62, 6388–6393 CrossRef CAS.
- (a) K. Ishihara, H. Ishibashi and H. Yamamoto, J. Am. Chem. Soc., 2001, 123, 1505–1506 CrossRef CAS; (b) K. Ishihara, S. Nakamura and H. Yamamoto, J. Am. Chem. Soc., 1999, 121, 4906–4907 CrossRef CAS; (c) S. Nakamura, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2000, 122, 8131–8140 CrossRef CAS.
- Y.-J. Zhao, S.-S. Chng and T.-P. Loh, J. Am. Chem. Soc., 2007, 129, 492–493 CrossRef CAS.
- (a) H. Imagawa, T. Iyenaga and M. Nishizawa, Synlett, 2005, 703–705 CAS; (b) H. Yamamoto, I. Sasaki, Y. Hirai, K. Namba, H. Imagawa and M. Nishizawa, Angew. Chem., Int. Ed., 2009, 48, 1244–1247 CrossRef CAS.
- S. G. Sethofer, T. Mayer and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8276–8277 CrossRef CAS.
- U. Hoffmann, Y. M. Gao, B. P. Pandey, S. Klinge, K. D. Warzecha, C. Kruger, H. D. Roth and M. Demuth, J. Am. Chem. Soc., 1993, 115, 10358–10359 CrossRef CAS.
- For a recent example of a tricyclization to produce “bent” polycyclic products from farnesyl aryl ethers and oxaterpenoids respectively, see: (a) R. A. Shenvi and E. J. Corey, Org. Lett., 2010, 12, 3548–3551 CrossRef CAS; (b) B. Li, Y.-C. Lai, Y. Zhao, Y.-H. Wong, Z.-L. Shen and T.-P. Loh, Angew. Chem., Int. Ed., 2012 Search PubMed , ASAP.
- Cyclization reactions have been observed when preceded by Claisen-rearrangement and termination by nucleophilic attack of oxygen. For an example, see ref. 7c.
- For cyclization reactions with termination by oxygen nucleophiles, see: (a) J. D. Neighbors, N. R. Mente, K. D. Boss, D. W. Zehnder and D. F. Wiemer, Tetrahedron Lett., 2008, 49, 516–519 CrossRef CAS; (b) J. D. Neighbors, J. A. Beutler and D. F. Wiemer, J. Org. Chem., 2005, 70, 925–931 CrossRef CAS; (c) C. A. Mullen and M. R. Gagne, J. Am. Chem. Soc., 2007, 129, 11880–11881 CrossRef CAS; (d) C. A. Mullen, A. N. Campbell and M. R. Gagne, Angew. Chem., Int. Ed., 2008, 47, 6011–6014 CrossRef CAS.
- For an example of an arylsulfonamide undergoing a Lewis acid-mediated cyclization reaction, see: G. Lemiere, B. Cacciuttolo, E. Belhassen and E. Dunach, Org. Lett., 2012, 14, 2750–2753 CrossRef CAS.
- For an example of a polycyclization reaction of an amide involving C–N bond formation, see: C. M. Haskins and D. W. Knight, Chem. Commun., 2005, 3162–3164 RSC.
- J. Barluenga, M. Trincado, E. Rubio and J. M. Gonzalez, J. Am. Chem. Soc., 2004, 126, 3416–3417 CrossRef CAS.
- For an earlier example of scandium triflate promoted cationic polycyclization, see: M. Bogenstatter, A. Limberg, L. E. Overman and A. L. Tomasi, J. Am. Chem. Soc., 1999, 121, 12206–12207 CrossRef.
- Although several strong chiral protic acids were also effective at promoting cyclization, no significant enantioselection was observed; BINBAM: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS; (b) A. Berkessel, P. Christ, N. Leconte, J.-M. Neudörfl and M. Schäfer, Eur. J. Org. Chem., 2010, 2010(27), 5165–5170 CrossRef; JINGLE: (c) P. Garcia-Garcia, F. Lay, P. Garcia-Garcia, C. Rabalakos and B. List, Angew. Chem., Int. Ed., 2009, 48, 4363–4366 CrossRef CAS; N-triflyl-BINOL-phosphoramide: (d) D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS; For a review on strong protic acid catalysis, see: (e) M. Rueping, B. J. Nachtsheim, W. Ieawsuwan and I. Atodiresei, Angew. Chem., Int. Ed., 2011, 50, 6706–6720 CrossRef CAS; (f) M. Treskow, J. Neudorfl and R. Giernoth, Eur. J. Org. Chem., 2009, 3693–3697 CrossRef CAS.
- (a) S. E. Denmark, D. Kalyani and W. R. Collins, J. Am. Chem. Soc., 2010, 132, 15752–15765 CrossRef CAS; (b) S. E. Denmark, D. J. P. Kornfilt and T. Vogler, J. Am. Chem. Soc., 2011, 133, 15308–15311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 880423 and 880424. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21405a |
| This journal is © The Royal Society of Chemistry 2013 |