Spacer length shapes drug release and therapeutic efficacy of traceless disulfide-linked ADCs targeting the tumor neovasculature†
Martina
Steiner
a,
Isabelle
Hartmann
a,
Elena
Perrino
a,
Giulio
Casi
b,
Samatanga
Brighton
cd,
Ilian
Jelesarov
c,
Gonçalo J. L.
Bernardes‡
*a and
Dario
Neri
a
aDepartment of Chemistry and Applied Biosciences, Swiss Federal Institute of Technology (ETH Zürich), Wolfgang-Pauli Str. 10, 8093 Zürich, Switzerland. E-mail: Goncalo.Bernardes@pharma.ethz.ch; Goncalo.Bernardes@linacre.oxon.org; Fax: +41 44633 1358
bPhilochem AG, Libernstrasse 3, 8112 Otelfingen, Switzerland
cDepartment of Biochemistry, University of Zürich, Winterthurerstr. 190, 8057 Zürich, Switzerland
dDepartment of Biology, Institute of Molecular Biology and Biophysics, Swiss Federal Institute of Technology (ETH Zürich), Schafmattstrasse 20, 8093 Zürich, Switzerland
First published on 8th October 2012
Abstract
We systematically investigated how the therapeutic efficacy of a traceless, vascular targeting antibody–drug conjugate (ADC) is affected by the length of a spacer introduced between the antibody's globular fold and the site of drug attachment. Homogeneous ADCs were prepared from the direct conjugation of engineered C-terminal cysteines with a potent thiol containing drug which was separated from the antibody surface by unstructured spacers of increasing length. We found that a smaller spacer length is reflected in enhanced stability and therapeutic efficacy of the conjugates in a syngeneic model of murine cancer.
Introduction
Antibody–drug conjugates (ADCs) exploit the ability of antibodies to specifically accumulate at sites of disease for the targeted delivery of potent cytotoxic drugs. ADCs can improve the therapeutic index of small molecule anticancer agents by modulating their distribution in the body, but also enhance the antitumor activity of unmodified, “naked” therapeutic antibodies by adding an additional effector payload.1–5 The concept of ADCs is compellingly simple, but they have only recently demonstrated a clear benefit for cancer patients.6–8 While these clinical successes represent a major hope in the fight against cancer, many aspects of ADC design including choice of conjugation chemistry, drug release strategy, antigen and antibody format remain not fully understood.ADCs are pro-drugs that require the release of the drug from the antibody vehicle. Release can either occur (i) directly in the tumor cell or (ii) indirectly in the local tumor environment. In the first case, internalization occurs after receptor-mediated endocytosis is triggered by the antibody binding to an internalizing receptor on the cell surface. The activation in target cells depends either on lysosomal degradation of the antibody,9,10 selective cleavage of the linker by lysosomal proteases,11 pH-dependent release when using acid-labile hydrazone containing linkers12 or disulfide containing linkers that are selectively cleaved in a reducing endosomal compartment.9,10,13 However, it has been shown that internalization is not an absolute requirement for drug-release from ADCs. Localised release of the drug in the tumor's extracellular environment can occur and successfully inflict damage to cancer cells, as demonstrated using ADCs targeting the mucin secreted by cells14 and antigens in the tumor neovasculature.15 Splice isoforms of tenascin-C and oncofetal fibronectin are highly expressed in the subendothelial extracellular matrix (ECM) during the formation of new blood vessels, a process essential for the growth of most solid tumors.16,17 Antibodies against these antigens have in the past shown highly selective accumulation in models of solid tumors in mice as well as in cancer patients.16 Thus, ADCs targeting these markers may, in principle, be used for the treatment of a wide range of aggressive solid cancers.17,18
Traditional drug conjugation strategies often yield heterogeneous ADC preparations with different drug to antibody ratios and pharmacokinetics.1–4 The use of site-specific protein modification methodologies has been used to improve batch-to-batch reproducibility and to enhance the therapeutic index of ADCs.15,19–21 A recent study has addressed the influence of the site and local charge of conjugation on the stability of ADCs.22 Engineered cysteines of trastuzumab were conjugated to a maleimide containing an auristatin derivative that is equipped with a protease cleavable site. The resulting thioether succinimide, often regarded as stable, may undergo retro and exchange reactions in the presence of nucleophiles.23,24 This adds an extra cleavable site from which the drug with the retained linker may be released. Both the site and local charge of the thioether succinimide linkage were demonstrated to have a dramatic effect on the stability and on the therapeutic index of the resulting ADCs.22 In a different study, it has also been demonstrated that the position of a DOTA chelator in an antibody is reflected on the biodistribution and targeting properties of the conjugate.25 Together, these examples highlight the need for innovative strategies that lead to homogeneous ADCs with a chemically defined linkage for drug release. Such conjugates would facilitate pharmacokinetic analysis and a clearer assessment of the therapeutic efficacy of ADCs.15,19
In the present work, we systematically studied the influence of the spacing between the globular fold of the antibody and the cytotoxic drug on drug release and therapeutic efficacy of chemically defined ADCs targeting the tumor neovasculature. Using site-specific modification chemistry, homogeneous disulfide-linked ADCs were produced differing in the length between the antibody surface and the cleavable disulfide. Key to our approach is the generation of traceless ADCs, that is, ADCs that possess a defined cleavable linkage that upon a certain trigger releases the free drug and native antibody. This enabled a clean dissection of structure activity relationships of vascular ADCs equipped with spacers of different size between antibody surface and modified cysteine. We found that a smaller spacer length from the globular fold of the antibody results in enhanced stability towards disulfide reduction and improved therapeutic efficacy in a syngeneic tumor model in immunocompetent mice.
Results and discussion
To analyze the effect of spacer length on drug-release, stability and therapeutic efficacy, we started by genetically engineering small immunoproteins (F8), SIP(F8), and antibody fragments with C-terminal cysteines which were separated from the globular fold by spacers of increasing length (2, 5 and 7 amino acid residues). Neutral amino acids without or with small side chains (combinations of glycine G and alanine A) were chosen to build unstructured spacers. The resulting antibodies were named according to their C-terminal sequence, SIP(F8)GGC, SIP(F8)GGAGGC and SIP(F8)GGAGAGGC (Fig. 1a). The F8 antibody is specific to the extracellular domain A (EDA) of fibronectin, a splice isoform associated with tumor angiogenesis.16,26 In addition, antibodies in the SIP format targeting fibronectin accumulate rapidly at the tumor soon after injection27 as demonstrated by quantitative biodistribution28 and by nuclear medicine studies.29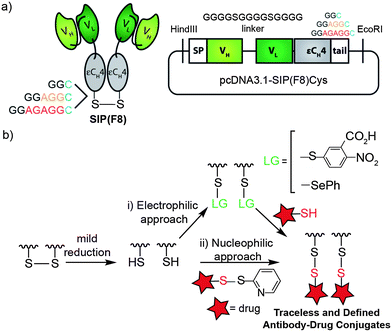 | ||
| Fig. 1 Construction of traceless, chemically defined tumor vascular targeting ADCs. (a) Schematic representation of the SIP antibody fragment and the domain arrangement when cloned in a pcDNA3.1 vector. Three variants were produced with 2, 5 or 7 extra amino acids before the C-terminal cysteine. The F8 antibody is specific to the EDA domain of human and murine fibronectin. (b) Illustration for the generation of homogenous mixed disulfide ADCs by (i) an electrophilic approach, where the cysteine on the antibody is activated with Ellman's reagent or phenylselenenyl bromide prior to conjugation with a thiol drug, and (ii) a nucleophilic approach, where the cysteine acts as a nucleophile. | ||
Traceless, disulfide-linked ADCs were produced using a site-specific conjugation strategy based on the direct modification of engineered C-terminal cysteines with a potent cytotoxic thiol analogue of cemadotin.15 Mild reduction of the C-terminal interchain disulfide bridge between the two subunits of the SIP antibody fragment using tris(2-carboxyethyl)phosphine (TCEP) results in one free cysteine per monomer that can act as an electrophile or as a nucleophile (Fig. 1b). In the electrophilic approach, the C-terminal cysteine is first exposed to either Ellman's reagent (5,5′-dithiobis-(2-nitrobenzoic acid)-DTNB)30–33 or phenylselenyl bromide34–37 followed by the addition of a thiol-containing cemadotin analogue (CemCH2-SH) to yield a homogeneous mixed disulfide-linked ADC. Addition of a disulfide pyridyl cemadotin analogue to the free C-terminal cysteine, which then acts as a nucleophile, can also be successfully used for the formation of a defined mixed disulfide conjugate (see the ESI† for details).§ Of these methods, the one where an antibody-Ellman's intermediate is formed revealed to be the most practical and robust. Conjugation reactions proceeded within minutes, with high conversion (>95%) and high yields as analyzed by ESI-MS (Fig. 2d). Importantly, we use as little as 10 equivalents of CemCH2-SH. The rapid formation of the mixed disulfide is driven by the low pKa of the aromatic thiol, which prevents the competing formation of a symmetric disulfide.
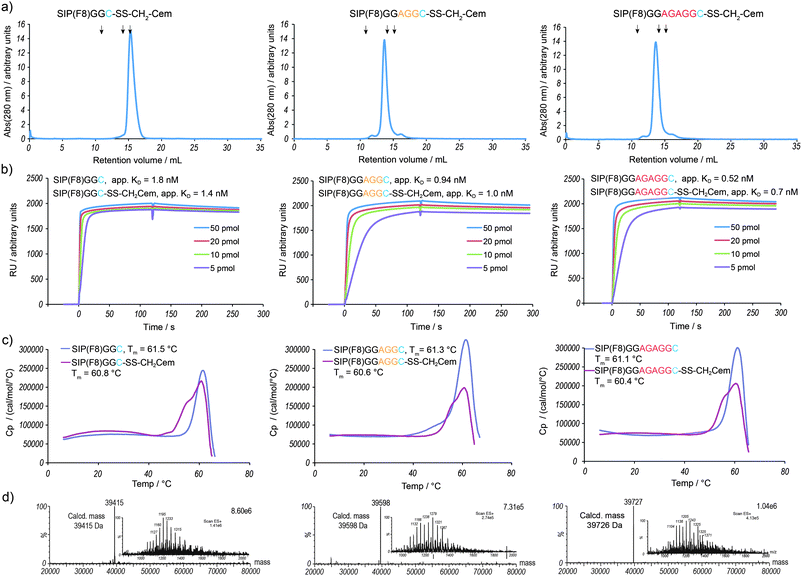 | ||
| Fig. 2 Analytical characterization of the vascular targeting ADC variants SIP(F8)GGC-SS-CH2Cem, SIP(F8)GGAGGC-SS-CH2Cem and SIP(F8)GGAGAGGC-SS-CH2Cem. (a) Gel-filtration analysis of purified ADCs. The peaks eluting at a retention volume of 14 to 15 mL correspond to the noncovalent homodimeric form of the ADCs. Arrows indicate standard proteins (11 mL: ferritin 440 kDa; 14.1 mL: BSA 67 kDa; 15.4 mL: β-lactoglobulin 35 kDa). (b) The apparent KD values were determined by Biacore analysis of purified ADCs towards recombinant 11A12 domains of fibronectin. (c) Differential scanning calorimetry of purified antibody and ADCs. The melting temperature of the conjugate is lower compared to the uncoupled protein. (d) LC-MS spectra of purified ADCs: deconvoluted and ion series (inset). | ||
The ADCs run as a single, homodimeric species in size-exclusion chromatography (Fig. 2a). The antigen-binding capacity of the ADCs (Fig. 2b) is retained when compared to the unconjugated antibody. The thermal stability of the three variants is identical, with the melting temperature Tm of the conjugate being 0.7 °C lower than the Tm of the unconjugated antibody (Fig. 2c). The use of typical conjugation strategies often results in conjugates with lower antigen binding capacity and decreased Tm when compared to naked antibodies.24,38 The conjugates were shown to be stable under storage conditions (−80 °C and 4 °C, see the ESI†). Our traceless and site-specific strategy produces mixed disulfide-linked ADCs with excellent chemical definition, a key feature for the development of cleaner ADCs.
Having in hand three disulfide-linked SIP(F8) drug conjugates that only differ in the length by which the drug is separated from the antibody surface, we next evaluated their reactivity in the presence of different physiological amounts of glutathione (GSH)39,40 (Fig. 3a and b). The ADCs reacted readily in the presence of GSH, with as little as 0.9 mM being sufficient to observe disulfide reduction after 10 minutes. By using a vascular targeting approach for the delivery of drugs to the tumor microenvironment, we expect small amounts of physiological reducing agents such as GSH or cysteine to be able to trigger disulfide bond reduction and the consequent release of the cytotoxic payload from the antibody. A longer length between antibody surface and modified cysteine enhances the disulfide reduction. For example, when SIP(F8)GGAGAGGC-SS-CH2Cem is incubated with 2.7 mM GSH, only 30% of the intact ADC is left after 10 min. Under the same conditions, SIP(F8)GGC-SS-CH2Cem remains 70% intact (Fig. 3a and b). The same trend was also observed when we measured the rate of release of Ellman's from the different SIP(F8)-Ellman's conjugates after incubation with different amounts of 1,4-dithio-D-threitol (DTT) (see the ESI† for details). Furthermore, we studied the stability of the different ADC constructs in plasma using ESI-MS (see the ESI†). ADCs were found to be essentially stable after 24 hours incubation in mouse plasma at 37 °C with SIP(F8)GGC-SS-CH2Cem remaining 85% intact under such conditions (see the ESI†). In addition, we also observed that conjugate stability in plasma inversely correlates with spacer length, i.e. a smaller spacer results in enhanced stability.
 | ||
| Fig. 3 Release of mixed disulfide ADCs studied in vitro. (a) Release of CemCH2-SH after 10 min incubation of ADC with different equivalents of glutathione (GSH). Percentage of intact ADC and free antibody was assessed by LC-MS. (b) Corresponding deconvoluted LC-MS spectra for the three ADC variants incubated at increasing concentrations of GSH. | ||
We next decided to analyze whether these differences in reactivity translate into a diverging therapeutic effect in mouse models of cancer. The three variants of SIP(F8) ADCs with distinct reactive disulfide profiles were then tested in an A20 lymphoma model (Fig. 4). Previous quantitative biodistribution studies in BALB/c mice bearing systemic A20 syngeneic lymphoma revealed a selective accumulation and retention of an SIP antibody targeting the extra-cellular domain of fibronectin,41 making this model particularly suitable to test the in vivo efficacy of our conjugates. Five injections of 43 mg kg−1 ADC on consecutive days lead to a considerable tumor growth retardation, curing one out of five mice in each therapy group without any detectable associated toxicity. Importantly, we found that the degree of tumor growth retardation correlates with the readiness of drug release. The ADC with the smaller spacer between the globular fold of the antibody and the drug revealed to be the most effective in vivo.
 | ||
| Fig. 4 Therapeutic efficacy of ADC variants in a syngeneic murine lymphoma model. (a) Immunocompetent female BALB/c mice bearing subcutaneous A20 reticulum cell sarcoma cells were treated intravenously with 43 mg kg−1 of SIP(F8)GGC-CH2Cem, SIP(F8)GGAGGC-CH2Cem and SIP(F8)GGAGAGGC-CH2Cem or saline (5 mice per group). Treatment was performed daily for a period of 5 days (arrows). Therapy was initiated when tumors reached a size of 80–120 mm3. Data represents mean tumor volumes (±standard error). Tumor growth curves were stopped when the tumors reached a size of 1500 mm3. (b) Body weight variations of the mice during and after therapy. No detectable weight loss was observed. (c) Survival curves of treatment and control groups; substantial prolongation for mice which received ADC treatment. One out of 5 mice in the therapy groups were cured. (d) Photos of mice 10 days after tumor implantation. | ||
Finally, we decided to further demonstrate the in vivo efficacy of our non-internalizing, traceless ADC approach for the delivery of cytotoxic drugs to the tumor microenvironment in a different murine model of cancer. As tumor model, we chose CT26 colon carcinoma where the F8 antibody has been shown to strongly react with neovascular structures in immunofluorescence analysis.42 We compared the naked antibody, the original cemadotin drug (Cem) and its thiol derivative (CemCH2-SH) with the tumor neovasculature targeting ADC bearing the smallest spacer, SIP(F8)GGC-SS-CH2Cem, and an ADC with an antibody against an irrelevant antigen (the anti-hen-egg-lysozyme antibody KSF). Only the administration of the anti-EDA ADC resulted in a potent reduction in tumor growth and a significant prolongation of life expectancy (Fig. 5). This result confirms the potential of using the tumor microenvironment for the efficient delivery of cytotoxic drugs for cancer therapy.15,43
 | ||
| Fig. 5 Therapeutic efficacy of SIP(F8)GGC-SS-CH2Cem in a syngeneic murine carcinoma model. (a) Immunocompetent female BALB/c mice bearing subcutaneous CT26 colon carcinoma cells were treated intravenously with 43 mg kg−1 of SIP(F8)GGC-CH2Cem, SIP(KSF)GGC-CH2Cem, SIP(F8)GGC, 0.75 mg kg−1 CemCH2-SH, Cem or saline (5 mice per group). Treatment was performed daily for a period of 5 days (arrows). Therapy was initiated when tumors reached a size of 80–120 mm3. Data represents mean tumor volumes (±standard error). Tumor growth curves were stopped when tumors reached a size of 800 mm3. (b) Body weight variations of the mice during and after therapy. No detectable weight loss was observed. (c) Survival curves of treatment and control groups; substantial prolongation for mice which received anti-EDA ADC treatment. Naked antibody, ADC against an irrelevant target, and the unconjugated drugs did not have a therapeutic benefit. *At day 24, 3 mice from the therapy group were sacrificed with tumor volumes between 300–400 mm3. (d) Photos of mice 9 days after tumor implantation. | ||
Conclusions
The use of site-specific protein modification has allowed us to generate traceless and chemically defined ADCs which only differ in the length of the spacer between the globular fold of the antibody and the drug. The mechanism by which the drug is released from the vascular targeting antibody at the site of action most likely relies on reducing agents in the tumor environment. Indeed, we were able to demonstrate the direct contribution of GSH on drug release in vitro. The variants where the disulfide linkage is less hindered, thus protruding from the globular antibody, are more prone to reduction and consequent drug release. At the tumor site, dying cells initially release additional GSH and cysteine, locally reinforcing the drug release and amplifying the drug-mediated cell killing.1,15 The destabilizing effect of placing additional amino acid residues between the antibody surface and cleavable disulfide is also reflected in the therapeutic outcome with therapeutic efficacy inversely correlating to the length of the spacer between CH4 domain and modified cysteine. We hypothesise that the inferior therapeutic activity observed for the ADC with a longer spacer stems from decreased stability.This work underscores the power of site-specific protein modification for the systematic study of structure activity relationships of ADCs. The ability to attach a drug at a precise site within the antibody resulting in a defined cleavable linkage for drug release is key for the development of ADCs. This may be achieved by using a strategy in which the new linkage formed from the conjugation reaction can be cleaved in vivo, as the disulfide described in this work, or by genetically44,45 or chemically46,47 introducing unnatural amino acids into the antibody with functionalities that may be used for site-specific modification and that results in a cleavable linkage for drug release.19 In addition, the concepts studied here provide important insights for the development of ADCs beyond oncology, as angiogenesis is also a common feature of diseases such as rheumatoid arthritis,48 endometriosis49 and atherosclerosis.50
Acknowledgements
We thank Dr Filipa P. da Cruz for her valuable help during therapy experiments and helpful comments on the manuscript. We also thank Dr Sabrina Trüssel for preliminary contributions on this project, and Nikolaus Krall for his helpful comments on the manuscript. This work was supported by ETH Zürich, Swiss National Science Foundation, SwissBridge/Stammbach Stiftung, Kommission für Technologie und Innovation (KTI) and Philochem AG and Philogen SpA. G.J.L.B. is an EMBO and Novartis Research Fellow.Notes and references
- G. Casi and D. Neri, J. Control. Release, 2012, 161, 422–428 CrossRef CAS.
- P. D. Senter, Curr. Opin. Chem. Biol., 2009, 13, 235–244 CrossRef CAS.
- R. V. J. Chari, Acc. Chem. Res., 2008, 41, 98–107 CrossRef CAS.
- S. C. Alley, N. M. Okeley and P. D. Senter, Curr. Opin. Chem. Biol., 2010, 14, 529–537 CrossRef CAS.
- A. M. Wu and P. D. Senter, Nat. Biotechnol., 2005, 23, 1137–1146 CrossRef CAS.
- P. D. Senter and E. L. Sievers, Nat. Biotechnol., 2012, 30, 631–637 CrossRef CAS.
- A. Younes, N. L. Bartlett, J. P. Leonard, D. A. Kennedy, C. M. Lynch, E. L. Sievers and A. Forero-Torres, N. Engl. J. Med., 2010, 363, 1812–1821 CrossRef CAS.
- H. A. Burris, 3rd, H. S. Rugo, S. J. Vukelja, C. L. Vogel, R. A. Borson, S. Limentani, E. Tan-Chiu, I. E. Krop, R. A. Michaelson, S. Girish, L. Amler, M. Zheng, Y. W. Chu, B. Klencke and J. A. O'Shaughnessy, J. Clin. Oncol., 2010, 29, 398–405 CrossRef.
- H. K. Erickson, P. U. Park, W. C. Widdison, Y. V. Kovtun, L. M. Garrett, K. Hoffman, R. J. Lutz, V. S. Goldmacher and W. A. Blättler, Cancer Res., 2006, 66, 4426–4433 CrossRef CAS.
- G. D. Lewis Phillips, G. Li, D. L. Dugger, L. M. Crocker, K. L. Parsons, E. Mai, W. A. Blättler, J. M. Lambert, R. V. J. Chari, R. J. Lutz, W. L. T. Wong, F. S. Jacobson, H. Koeppen, R. H. Schwall, S. R. Kenkare-Mitra, S. D. Spencer and M. X. Sliwkowski, Cancer Res., 2008, 68, 9280–9290 CrossRef CAS.
- S. O. Doronina, B. E. Toki, M. Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS.
- P. Trail, D. Willner, S. Lasch, A. Henderson, S. Hofstead, A. Casazza, R. Firestone, I. Hellstrom and K. Hellstrom, Science, 1993, 261, 212–215 CAS.
- B. A. Kellogg, L. Garrett, Y. Kovtun, K. C. Lai, B. Leece, M. Miller, G. Payne, R. Steeves, K. R. Whiteman, W. Widdison, H. Xie, R. Singh, R. V. Chari, J. M. Lambert and R. J. Lutz, Bioconjugate Chem., 2011, 22, 717–727 CrossRef CAS.
- R. M. Sharkey, H. Karacay, S. V. Govindan and D. M. Goldenberg, Mol. Cancer Ther., 2011, 10, 1072–1081 CrossRef CAS.
- G. J. L. Bernardes, G. Casi, S. Trüssel, I. Hartmann, K. Schwager, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2012, 51, 941–944 CrossRef CAS.
- D. Neri and R. Bicknell, Nat. Rev. Cancer, 2005, 5, 436–446 CrossRef CAS.
- J.-N. Rybak, C. Roesli, M. Kaspar, A. Villa and D. Neri, Cancer Res., 2007, 67, 10948–10957 CrossRef CAS.
- H. P. Gerber, P. D. Senter and I. S. Grewal, mAbs, 2009, 1, 247–253 CrossRef.
- G. Casi, N. Huguenin-Dezot, K. Zuberbühler, J. Scheuermann and D. Neri, J. Am. Chem. Soc., 2012, 134, 5887–5892 CrossRef CAS.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS.
- K. Zuberbühler, G. Casi, G. J. L. Bernardes and D. Neri, Chem. Commun., 2012, 48, 7100–7102 RSC.
- B. Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S. F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS.
- M. Acchione, H. Kwon, C. M. Jochheim and W. M. Atkins, mAbs, 2012, 4, 362–372 CrossRef.
- A. Perols, H. Honarvar, J. Strand, R. Selvaraju, A. Orlova, A. Eriksson Karlström and V. Tolmachev, Bioconjugate Chem., 2012, 23, 1661–1670 CrossRef CAS.
- A. Villa, E. Trachsel, M. Kaspar, C. Schliemann, R. Sommavilla, J. N. Rybak, C. Rösli, L. Borsi and D. Neri, Int. J. Cancer, 2008, 122, 2405–2413 CrossRef CAS.
- L. Borsi, E. Balza, M. Bestagno, P. Castellani, B. Carnemolla, A. Biro, A. Leprini, J. Sepulveda, O. Burrone, D. Neri and L. Zardi, Int. J. Cancer, 2002, 102, 75–85 CrossRef CAS.
- D. Berndorff, S. Borkowski, S. Sieger, A. Rother, M. Friebe, F. Viti, C. S. Hilger, J. E. Cyr and L. M. Dinkelborg, Clin. Cancer Res., 2005, 11, 7053s–7063s CrossRef CAS.
- S. Sauer, P. A. Erba, M. Petrini, A. Menrad, L. Giovannoni, C. Grana, B. Hirsch, L. Zardi, G. Paganelli, G. Mariani, D. Neri, H. Durkop and H. D. Menssen, Blood, 2009, 113, 2265–2274 CrossRef CAS.
- C. Chatterjee, R. K. McGinty, B. Fierz and T. W. Muir, Nat. Chem. Biol., 2010, 6, 267–269 CrossRef CAS.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. E. Oualid, T. D. W. Claridge and B. G. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS.
- G. J. L. Bernardes, D. P. Gamblin and B. G. Davis, Angew. Chem., Int. Ed., 2006, 45, 4007–4011 CrossRef CAS.
- D. P. Gamblin, P. Garnier, S. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 43, 828–833 CrossRef CAS.
- D. P. Gamblin, S. van Kasteren, G. J. L. Bernardes, J. M. Chalker, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Mol. BioSyst., 2008, 4, 558–561 RSC.
- O. Boutureira, G. J. L. Bernardes, M. Fernández-González, D. C. Anthony and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1432–1436 CrossRef CAS.
- A. A. Wakankar, M. B. Feeney, J. Rivera, Y. Chen, M. Kim, V. K. Sharma and Y. J. Wang, Bioconjugate Chem., 2010, 21, 1588–1595 CrossRef CAS.
- A. Meister and M. E. Anderson, Annu. Rev. Biochem., 1983, 52, 711–760 CrossRef CAS.
- C. Wu, C. Belenda, J.-C. Leroux and M. A. Gauthier, Chem.–Eur. J., 2011, 17, 10064–10070 CrossRef CAS.
- C. Schliemann, A. Palumbo, K. Zuberbühler, A. Villa, M. Kaspar, E. Trachsel, W. Klapper, H. D. Menssen and D. Neri, Blood, 2009, 113, 2275–2283 CrossRef CAS.
- N. Pasche, S. Wulhfard, F. Pretto, E. Carugati and D. Neri, Clin. Cancer Res., 2012, 18, 4092–4103 CrossRef CAS.
- A. Palumbo, F. Hauler, P. Dziunycz, K. Schwager, A. Soltermann, F. Pretto, C. Alonso, G. F. Hofbauer, R. W. Boyle and D. Neri, Br. J. Cancer, 2011, 104, 1106–1115 CrossRef CAS.
- J. Xie and P. G. Schultz, Nat. Rev. Mol. Cell Biol., 2006, 7, 775–782 CrossRef CAS.
- L. Davis and J. W. Chin, Nat. Rev. Mol. Cell Biol., 2012, 13, 168–182 CAS.
- J. M. Chalker, G. J. L. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS.
- J. M. Chalker and B. G. Davis, Curr. Opin. Chem. Biol., 2010, 14, 781–789 CrossRef CAS.
- E. Trachsel, F. Bootz, M. Silacci, M. Kaspar, H. Kosmehl and D. Neri, Arthritis Res. Ther., 2007, 9, R9 Search PubMed.
- K. Schwager, F. Bootz, P. Imesch, M. Kaspar, E. Trachsel and D. Neri, Hum. Reprod., 2011, 26, 2344–2352 CrossRef CAS.
- M. Fiechter, K. Frey, T. Fugmann, P. A. Kaufmann and D. Neri, Atherosclerosis, 2011, 214, 325–330 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details including 1H NMR, MS and HPLC data for all drug derivatives, cloning, expression and characterization of antibodies, and ESI-MS, gel filtration, SDS-PAGE, DSC, and Biacore for all antibody–drug conjugates. See DOI: 10.1039/c2sc21107f |
| ‡ Present address: Instituto de Medicina Molecular, Faculdade de Medicina, 1649-028 Lisboa, Portugal. |
| § These approaches for the formation of mixed disulfide-linked ADCs, were also used successfully for the construction of diabody(F8)–GGC conjugates. See the ESI† for details. |
| This journal is © The Royal Society of Chemistry 2013 |