Nonheme oxoiron(IV) complexes of pentadentate N5 ligands: spectroscopy, electrochemistry, and oxidative reactivity†
Dong
Wang
a,
Kallol
Ray‡
a,
Michael J.
Collins
ab,
Erik R.
Farquhar
a,
Jonathan R.
Frisch
a,
Laura
Gómez
c,
Timothy A.
Jackson
a,
Marion
Kerscher
d,
Arkadius
Waleska
d,
Peter
Comba
*d,
Miquel
Costas
*c and
Lawrence
Que
Jr.
*a
aDepartment of Chemistry and Center for Metals in Biocatalysis, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455, USA. E-mail: larryque@chem.umn.edu
bDepartment of Chemistry, Viterbo University, La Crosse, WI 54601, USA
cDepartament de Química, Universitat de Girona, Campus de Montilivi, E-17071 Girona, Spain. E-mail: miquel.costas@udg.edu
dUniversität Heidelberg, Anorganisch-Chemisches Institut, INF 270, D-69120 Heidelberg, Germany. E-mail: peter.comba@aci.uni-heidelberg.de
First published on 25th October 2012
Abstract
Oxoiron(IV) species have been found to act as the oxidants in the catalytic cycles of several mononuclear nonheme iron enzymes that activate dioxygen. To gain insight into the factors that govern the oxidative reactivity of such complexes, a series of five synthetic S = 1 [FeIV(O)(LN5)]2+ complexes has been characterized with respect to their spectroscopic and electrochemical properties as well as their relative abilities to carry out oxo transfer and hydrogen atom abstraction. The Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O units in these five complexes are supported by neutral pentadentate ligands having a combination of pyridine and tertiary amine donors but with different ligand frameworks. Characterization of the five complexes by X-ray absorption spectroscopy reveals FeO bonds of ca. 1.65 Å in length that give rise to the intense 1s → 3d pre-edge features indicative of iron centers with substantial deviation from centrosymmetry. Resonance Raman studies show that the five complexes exhibit ν(FeO) modes at 825–841 cm−1. Spectropotentiometric experiments in acetonitrile with 0.1 M water reveal that the supporting pentadentate ligands modulate the E1/2(IV/III) redox potentials with values ranging from 0.83 to 1.23 V vs. Fc, providing the first electrochemical determination of the E1/2(IV/III) redox potentials for a series of oxoiron(IV) complexes. The 0.4 V difference in potential may arise from differences in the relative number of pyridine and tertiary amine donors on the LN5 ligand and in the orientations of the pyridine donors relative to the FeO bond that are enforced by the ligand architecture. The rates of oxo-atom transfer (OAT) to thioanisole correlate linearly with the increase in the redox potentials, reflecting the relative electrophilicities of the oxoiron(IV) units. However this linear relationship does not extend to the rates of hydrogen-atom transfer (HAT) from 1,3-cyclohexadiene (CHD), 9,10-dihydroanthracene (DHA), and benzyl alcohol, suggesting that the HAT reactions are not governed by thermodynamics alone. This study represents the first investigation to compare the electrochemical and oxidative properties of a series of S = 1 FeIVO complexes with different ligand frameworks and sheds some light on the complexities of the reactivity of the oxoiron(IV) unit.
O units in these five complexes are supported by neutral pentadentate ligands having a combination of pyridine and tertiary amine donors but with different ligand frameworks. Characterization of the five complexes by X-ray absorption spectroscopy reveals FeO bonds of ca. 1.65 Å in length that give rise to the intense 1s → 3d pre-edge features indicative of iron centers with substantial deviation from centrosymmetry. Resonance Raman studies show that the five complexes exhibit ν(FeO) modes at 825–841 cm−1. Spectropotentiometric experiments in acetonitrile with 0.1 M water reveal that the supporting pentadentate ligands modulate the E1/2(IV/III) redox potentials with values ranging from 0.83 to 1.23 V vs. Fc, providing the first electrochemical determination of the E1/2(IV/III) redox potentials for a series of oxoiron(IV) complexes. The 0.4 V difference in potential may arise from differences in the relative number of pyridine and tertiary amine donors on the LN5 ligand and in the orientations of the pyridine donors relative to the FeO bond that are enforced by the ligand architecture. The rates of oxo-atom transfer (OAT) to thioanisole correlate linearly with the increase in the redox potentials, reflecting the relative electrophilicities of the oxoiron(IV) units. However this linear relationship does not extend to the rates of hydrogen-atom transfer (HAT) from 1,3-cyclohexadiene (CHD), 9,10-dihydroanthracene (DHA), and benzyl alcohol, suggesting that the HAT reactions are not governed by thermodynamics alone. This study represents the first investigation to compare the electrochemical and oxidative properties of a series of S = 1 FeIVO complexes with different ligand frameworks and sheds some light on the complexities of the reactivity of the oxoiron(IV) unit.
Introduction
High valent iron-oxo intermediates are considered to be the active species in the catalytic cycles of mononuclear nonheme iron enzymes that activate dioxygen.1–3 Within the last ten years, such species have been identified by rapid-freeze-quench techniques in several 2-oxoglutarate-dependent enzymes, namely TauD,4–7 prolyl 4-hydroxylase,8 and the halogenases CytC3 (ref. 9) and SyrB2,10 as well as the pterin-dependent phenylalanine11 and tyrosine hydroxylases.12 These intermediates have been characterized by Mössbauer spectroscopy as having high-spin iron(IV) centers. Furthermore, the presence of a terminal FeO unit in the TauD,6,7 CytC3,13 and SyrB2 (ref. 10) intermediates has been established by resonance Raman and/or Extended X-ray Absorption Fine Structure (EXAFS) methods.
Recent biomimetic efforts have identified a large number of mononuclear nonheme oxoiron(IV) complexes.14–17 Most of these complexes have been spectroscopically characterized to have S = 1 ground states and the FeIVO units are supported by a variety of polydentate ligand frameworks with mainly N donors, including macrocyclic cyclam,18–22 bicyclic bispidine,23–26 and tripodal ligand motifs.27–29 Only a few complexes have been shown experimentally to have S = 2 spin states.30–34 Crystal structures of five complexes have now been reported, namely [FeIV(O)(TMC)(NCCH3)]2+,19 [FeIV(O)(TMC-py)]2+,35 [FeIV(O)(N4Py)]2+ (1),36 [FeIV(O)(TMG3tren)]2+,37 and [FeIV(O)(H3buea)]− (ref. 32) (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane; TMC-py = 1,4,8-trimethyl-11-(2′-pyridylmethyl)-1,4,8,11-tetraaza-cyclotetradecane; TMG3tren = 1,1,1-tris{2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl}amine); H3buea, 1,1,1-tris[(N′-tert-butylureaylato)-N-ethyl]amine trianion; for N4Py, see Scheme 1). With this range of complexes, a question of significant interest is how the reactivity of the FeIVO unit is modulated by the various ligands that support it. For example, [FeIV(O)(TMC)(NCCH3)]2+ can only oxidize substrates with C–H bond dissociation energies (BDE) of <80 kcal mol−1, but replacing the axial CH3CN ligand with anions increases the rates of these reactions.38 On the other hand, some oxoiron(IV) complexes with combinations of N-heterocycle and amine donors have been found to attack even the strong C–H bonds of cyclohexane.28,29,39–41 For several of these complexes, an inverse linear correlation was obtained when log k′2 values (the second order rate constant normalized on per hydrogen atom basis) were plotted versus the substrate C–H bond strength, suggesting that the reactions proceed by hydrogen atom transfer; furthermore deuterium kinetic isotope effects (KIEs) much larger than the semiclassical limit were observed. Characterizing the redox properties of these synthetic complexes and correlating them with their reactivity towards substrates would thus be of great interest, and the present paper focuses on 1–5 (Scheme 1), a series of oxoiron(IV) complexes with pentadentate supporting ligands.
![Structures of oxoiron(iv) complexes 1–5 studied in this work. Numerical designations 1a–5a refer to corresponding [FeII(LN5)(NCCH3)]2+ complexes, while 1b–5b refer to corresponding [FeIII(OH)(LN5)]2+ species.](/image/article/2013/SC/c2sc21318d/c2sc21318d-s1.gif) | ||
| Scheme 1 Structures of oxoiron(IV) complexes 1–5 studied in this work. Numerical designations 1a–5a refer to corresponding [FeII(LN5)(NCCH3)]2+ complexes, while 1b–5b refer to corresponding [FeIII(OH)(LN5)]2+ species. | ||
The determination of the FeIV/III reduction potentials for the oxoiron(IV) complexes can provide quantitative insight into the thermodynamic factors that govern their differing oxidizing abilities. However, the electrochemical behavior of these complexes exhibits some complexity that has not been straightforward to interpret. For example, in the cyclic voltammetry (CV) of 1 in dry MeCN, the cathodic scan exhibited a wave at −0.44 V vs. Fc+/0,42 presumably reflecting the reduction of 1, while the anodic scan showed multiple poorly defined peaks of much lower intensity, suggesting the formation of a mixture of species upon reduction.43 The Ep,c value observed for 1 is in fact more negative than the E1/2 values associated with the FeIII/II couples of the corresponding iron(II) complexes [FeII(N4Py)(NCCH3)]2+ (E1/2 = 0.61 V vs. Fc+/0) and [FeII(N4Py)(OH2)]2+ (Ep,c = 0.15 V vs. Fc+/0)44–46,85 and would appear incommensurate with the reported ability of 1 to oxidize cyclohexane.39,47 This apparent contradiction led us to investigate the electrochemical properties of 1 under different conditions.
The irreversibility of the electrochemistry of 1 in CH3CN may derive from the expected high basicity of its one-electron reduced counterpart and its instability in the absence of a proton donor. Indeed there is only one well characterized example of an oxoiron(III) complex to date,48 and the oxide ligand in this complex is stabilized by several hydrogen bonding groups designed into the polydentate supporting ligand. Moreover, similar complications have been reported in the electrochemistry of RuIVO complexes and reversible cyclic voltammetric waves for the RuIV/III couples can be observed only in acidic aqueous solution.49–54 To circumvent this likely problem, we carried out electrochemical experiments in ‘wet’ CH3CN (CH3CN with added water) and used spectropotentiometry to elicit the Nernstian oxidation of the FeIII–OH complex 1b to 1 at a significantly more positive potential (E1/2(IV/III)) of 0.90 V vs. Fc+/0.46 In this work, we report a detailed comparison of complexes 1–5 with respect to their spectroscopic and electrochemical properties, and compare these properties with their rates of oxo-atom transfer (OAT) and hydrogen-atom transfer (HAT) to gain insights into what factors affect the reactivity of the FeIVO unit.
Results and discussion
A. Spectroscopic characterization of oxoiron(IV) complexes
Oxoiron(IV) complexes 1–5 of the pentadentate LN5 ligands shown in Scheme 1 were obtained by treatment of the iron(II) precursors 1a–5a with excess solid PhIO in CH3CN at 25 °C. After maximum formation of 1–5 (monitored by UV-vis spectroscopy), excess PhIO was removed by filtration. These complexes exhibit pale green or yellowish-green chromophores that derive from near-IR bands arising from ligand field transitions of an S = 1 FeIVO center (Table 1, Fig. 1),23,24,39 as assigned for 1 and [FeIV(O)(TMC)(NCCH3)]2+ on the basis of a detailed magnetic circular dichroism (MCD) analysis.55,56 A similar near-IR chromophore is observed for 3 (Fig. 1), which is reported for the first time in this study, and its formation is supported by the appearance of a dominant ion at m/z 546 in its electropspray ionization mass spectrometry (ESI-MS) spectrum with the isotope distribution pattern predicted for the formulation {[Fe(O)(Me2TACN-Py2)](CF3SO3)}+ (Fig. S1†).
| λ max (nm) in MeCN (ε, M−1 cm−1) |
ν(FeO) (cm−1) [Δ18O] |
XAS sample purity | XAS pre-edge | XAS edge energy (eV) | r(Fe–X)f (Å) | ||
|---|---|---|---|---|---|---|---|
| Area (normalized) | Energy (eV) | ||||||
| a From ref. 39. b Molar extinction coefficients estimated from thioanisole oxidation data. c From ref. 23. d From ref. 24. e Value obtained from the midpoint of a Fermi doublet at 831 and 847 cm−1 for the 16O sample. f EXAFS analysis of 2 reported in ref. 57; fits for the other complexes listed in Table S1.1 The typical uncertainty for first-shell distances by EXAFS analysis is ±0.02 Å. | |||||||
| 1 | 696 (400)a | 841 [−35] | 90% | 31 (34) | 7114.4 | 7124.0 | 1O @ 1.64 4N @ 1.96 |
| 2 | 739 (400), 900 (sh)a | 835 [−39] | 90% | 29 (32) | 7114.1 | 7123.7 | 1O @ 1.67 4N @ 2.00 |
| 3 | 740 (340), 900 (200)b | 839e [−35] | 85% | 29 (34) | 7114.1 | 7124.7 | 1O @ 1.63 4N @ 2.00 |
| 4 | 730 (400), 916 (sh)c,d | 840 [−35] | 74% | 24 (32) | 7114.5 | 7124.4 | 1O @ 1.64 3N @ 1.98 2N @ 2.15 |
| 5 | 730 (380), 896 (sh)c | 825 [−34] | 85% | 38 (45) | 7114.0 | 7123.4 | 1O @ 1.62 4N @ 1.97 1N @ 2.18 |
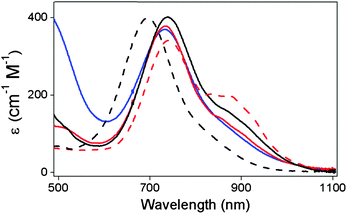 | ||
| Fig. 1 Electronic spectra of oxoiron(IV) complexes 1 (black dashed line), 2 (black solid line), 3 (red dashed line), 4 (red solid line) and 5 (blue solid line) recorded in CH3CN at room temperature. | ||
Table 1 also lists resonance Raman data for 1–5. These complexes exhibit a Raman feature at ca. 830 cm−1 upon 406.7 nm excitation that can be assigned to the FeO stretching mode on the basis of the large 18O shift elicited upon oxygen atom exchange with H218O. The ν(FeO)'s range from 841 cm−1 for 1 to 825 cm−1 for 5, with observed 18O downshifts fully consistent with that expected for a diatomic FeO oscillator.
X-ray absorption spectroscopic (XAS) data collected for 1–5 are also compiled in Table 1. All five complexes exhibit first inflection points in the rising Fe K-edge at 7124(1) eV, consistent with those obtained for other S = 1 oxoiron(IV) complexes in earlier studies.21,57,58 A single prominent pre-edge transition centered at 7114.2 ± 0.3 eV is observed for each complex, which is assigned to a 1s → 3d transition. This feature has a normalized pre-edge intensity of 32–34 units for 1–4 and 45 units for 5. The much larger pre-edge intensity of 5 presumably reflects a greater distortion from centrosymmetry at the iron center of 5 relative to 1–4, as suggested by density functional theory (DFT) calculations.59
EXAFS analysis of 1, 3, 4, and 5 (Fig. 2 and Tables 1 and S1†) reveals a short Fe–O distance at 1.62–1.64 Å associated with the FeO unit, which is comparable in length to the FeO distances found in the crystal structures of complexes 1 (1.639 Å),36 [FeIV(O)(TMC)(NCCH3)]2+ (1.646 Å),19 [FeIV(O)(TMC-py)]2+ (1.667 Å),35 [FeIV(O)(TMG3tren)]2+ (1.661 Å),37 and [FeIV(O)(H3buea)]− (1.680 Å).32 In addition, 1 and 3 exhibit a shell of nitrogen scatterers at 1.96 and 2.00 Å, respectively, assignable to ligated pyridine and tertiary amine donors. On the other hand, fits to 4 and 5 require two subshells of N scatterers at ca. 1.98 and 2.17 Å, consistent with structures derived from DFT calculations for 4 and 5 and reflecting the constraints imposed by the bispidine ligand framework.59
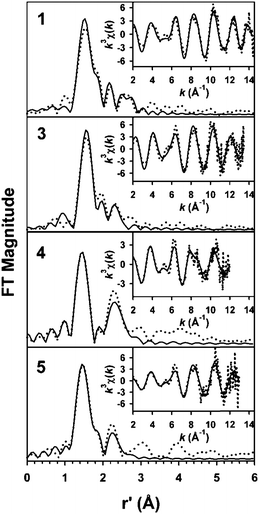 | ||
| Fig. 2 Unfiltered EXAFS spectra (dotted lines) and corresponding best fits (solid lines) of 1, 3, 4, and 5. The fits shown are given in bold italics in Table S1 in the ESI.† | ||
The data for the series of [FeIV(O)(LN5)] complexes presented above show that their spectroscopic properties are modulated by the nature of the supporting pentadentate ligand. The UV-vis spectra of all five complexes exhibit a principal feature at 696–740 nm, with a weaker band near 900 nm for 2–5 (Table 1). When the near-IR band of [FeIV(O)(TMC)(NCCH3)]2+ (λmax 824 nm) is included in the series, there is a trend in the absorbance maxima that reflects the length of the equatorial Fe–N bonds. Complex 1, with a 4-pyridine equatorial ligand set, exhibits a band at 696 nm at the low end of the series and has an average Fe–Neq bond length of 1.96 Å,36 while [FeIV(O)(TMC)(NCCH3)]2+, with a 4-tertiary-amine ligand set, has a band at 824 nm at the high end of the range and has an average Fe–Neq bond length of 2.09 Å.19 Complexes 2–5 have combinations of equatorial pyridine and tertiary amine ligands and average Fe–Neq bonds of intermediate lengths; consequently, their principal near IR features (740–745 nm) fall in between the two extremes. Therefore, the longer the Fe–N bond becomes, the weaker of an effect of the N donor can exert on the d-orbital splitting, the lower the energy of the ligand field transitions.
The accumulated characterization data suggest that 5 is the most distinct in properties of the five complexes presented here. While the EXAFS fits reported here do not reveal any significant differences among the five complexes, the X-ray absorption near edge structure (XANES)'data in Table 1 show that 5 has a pre-edge area much larger than those of the others (45 normalized units versus 32–34), indicating that it has a geometry with the largest distortion from centrosymmetry. In addition, Table 1 shows that 5 has a ν(FeO) value of 825 cm−1, which is lower than the 835 cm−1 value found for 2 and the ∼840 cm−1 value associated with 1, 3, and 4, suggesting that 5 has a somewhat weaker FeO bond than the rest. As will be seen in the subsequent sections, these differences can be related to our observations on their electrochemical potentials and oxidative reactivity.
B. Electrochemistry
To complement the spectroscopic results presented in the preceding section, electrochemistry experiments were carried out on 1–5 to assess their redox properties. As previously reported, cyclic voltammetric experiments on 1 carried out in dry CH3CN showed irreversible behavior, where a reduction wave (Ep,c) was observed only at a rather negative potential of −0.53 V vs. Fc+/0 at 25 °C with no accompanying feature in the oxidative scan.42,43,46,60 Analogous observations have also been previously described for [FeIV(O)(TMC)(NCCH3)]2+ by Nam and Fukuzumi.42,43 The reduction wave was assigned to the FeIV(O)/FeIII(O) reduction and its electrochemically irreversible nature was demonstrated to arise from slow electron transfer. For 2–5, irreversible CV behavior was observed in CH3CN as well, with all complexes showing broad reduction waves at similarly negative potentials (Table 2, Fig. S2†). Under identical conditions, the Ep,c values of the complexes were found to fall in the range of −0.30 to −0.60 V vs. Fc+/0 and decrease in the following order: 5 > 2 > 4 > 1 > 3.| Complex | E 1/2(III/II) (ΔE) in dry CH3CNb (V) | E p,c observed in dry CH3CNc (V) | E 1/2(IV/III) in CH3CN with 0.1 M waterd (V) |
|---|---|---|---|
| a All potentials are referred to Fc+/0. b Obtained by cyclic voltammetry with a scan rate = 0.1 V s−1 for 1 mM solutions of 1a–5a. c Obtained by cyclic voltammetry with a scan rate = 0.1 V s−1 for 1 mM solutions of 1–5. d Obtained by spectropotentiometry. e Estimated based on the correlation shown in Fig. 4. | |||
| 1 | 0.61 (0.08) | −0.53 | 0.90 |
| 2 | 0.69 (0.08) | −0.43 | 1.07 |
| 3 | 0.55 (0.07) | −0.58 | 0.83 |
| 4 | 0.72 (0.11) | −0.48 | 0.98e |
| 5 | 0.77 (0.14) | −0.32 | 1.23 |
We also tried to obtain the FeIV/III potentials of the complexes by electrochemical oxidation of iron(II) precursors 1a–5a in CH3CN. In dry solvent, oxidation of the iron(II) complexes gave rise to quasi-reversible CVs (Fig. S3†), with E1/2 values that did not depend significantly on scan rate (0.01–0.25 V s−1) or temperature. These features were found by coulometry to result from one-electron processes corresponding to the [Fe(LN5)(NCCH3)]3+/2+ couples, as previously reported for 1a.46 Further scans to higher potentials up to +1.25 V under the dry solvent conditions did not elicit any other features in the CVs that could be correlated to the formation of oxoiron(IV) species. This result should not be surprising, as there is no source of the oxygen atom that is needed to stabilize the iron(IV) oxidation state under the solvent conditions used. Subsequent cyclic voltammetric experiments carried out on 1a in CH3CN solutions containing 0.1 M H2O did not change the outcome, which is likely due to slow ligand exchange between bound solvent and water at the iron(III) stage relative to the CV time scale (Scheme 2).46,60,85
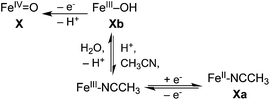 | ||
| Scheme 2 | ||
However high yields of FeIVO complexes 2, 3, and 5 could be obtained by bulk electrolytic oxidation of 2a, 3a, and 5a at applied potentials of ≥1.3 V in CH3CN with added water. The rate of oxidation depended on the concentration of water, and no electrochemical generation of the oxoiron(IV) species occurred in dry CH3CN. The bulk electrolytic oxidation required 30 minutes to complete when carried out in CH3CN with 0.1 M water. For each complex, chronocoulometry gave approximately the number of coulombs (within 5–10%) expected for a two-electron redox reaction. On the other hand, for some undetermined reason, bulk electrolysis of 4a only afforded a 40% yield of 4.
The bulk electrolytic oxidation could be followed spectrophotometrically in a spectroelectrochemical cell (Fig. 3). Like previous results reported for 1,46 these experiments revealed a two-step process to generate the oxoiron(IV) complex. In the first step, an intense near-UV band was generated around 350 nm,46 a chromophore we assign to the hydroxo-to-iron(III) charge transfer transition of the corresponding FeIII–OH complex derived from the one-electron oxidation of the iron(II) precursor.44,61–65 Electron paramagnetic resonance (EPR) studies of chemically oxidized (with tris(4-bromophenyl)aminium perchlorate) solutions of 1a–5a under the same solvent conditions showed signals associated with the formation of high-spin iron(III) species (Fig. S4†). In the second step of the bulk electrolytic oxidation, the near-UV chromophore of the FeIII–OH complex decayed concomitant with the appearance of the characteristic near-IR band of the oxoiron(IV) complex, and isosbestic points were observed for this transformation (Fig. 3).
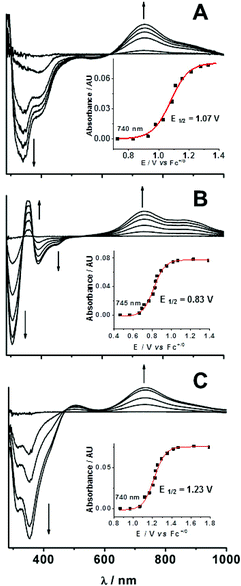 | ||
| Fig. 3 Difference spectra derived from spectropotentiometric titrations for complexes 2 (A), 3 (B), and 5 (C) at 25 °C in CH3CN containing 0.1 M H2O. The iron concentration used for all three experiments was 0.25 mM. For each panel, the increase in the absorbance at ∼740 nm is plotted as a function of the applied potential to show a sigmoidal shape that can be fit well with the Nernst equation. | ||
The conversion of [FeIII(OH)(LN5)]2+ species to [FeIV(O)(LN5)]2+ was then monitored in a stepwise fashion as a function of applied potential. Difference spectra vs. those of the [FeIII(OH)(LN5)]2+ complexes were calculated, and representative spectra for the formation of 2, 3 and 5 are shown in Fig. 3. A plot of the absorbance at 740 nm for 2 as a function of the applied potential shown in Fig. 3A inset shows a sigmoidal shape that can be fit well with the Nernst equation to give a midpoint potential E1/2 of +1.07(1) V vs. Fc+/0 for the FeIV/III couple. Similarly good fits to corresponding data for 3 (Fig. 3B inset) and 5 (Fig. 3C inset) gave respective E1/2 values of +0.83(1) V and +1.23(1) V vs. Fc+/0. The corresponding experiment for 4 proved unsuccessful, as it was not possible to obtain 4 in high yield by electrochemical oxidation. For 1, 2, 3, and 5, the E1/2(IV/III) values were independent of the concentration of H2O (up to 5 M) added into the CH3CN solutions.
In contrast, bulk electrolytic reduction under the same solvent conditions takes a much longer time at any given applied potential than the corresponding oxidative process. This illustrates the complexity in the electrochemical behavior of nonheme FeIVO complexes that has been observed previously but is not well understood42,43,46,60,66 and may be related to the requirement for a proton in the reduction (Scheme 2). Similar difficulties have been reported in the electrochemistry of RuIVO complexes in organic solvents, which were alleviated in aqueous solution.49,50,54,67,68 Despite these electrochemical complexities, the spectropotentiometric information we have obtained for a series of FeIVO complexes in this work nevertheless provides quite useful chemical insight (vide infra).
The FeIV/III potentials measured by spectropotentiometry for the four complexes are over 1.2 V more positive than corresponding Ep,c values described above in CV experiments in dry acetonitrile. Nevertheless, they exhibit the same decreasing order: 5 > 2 > 1 > 3. In fact, when the E1/2(IV/III) values obtained by spectropotentiometry are plotted against the Ep,c values obtained by CV, a linear correlation can be observed (Fig. 4, filled circles), demonstrating that both measurements are sensitive to the same ligand-specific effects on the potential. This plot thus allows us to estimate the E1/2(IV/III) value of 4 to be +0.98 V based on its Ep,c value measured in dry CH3CN.
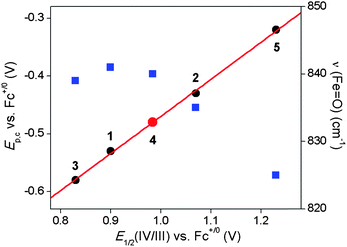 | ||
| Fig. 4 Plot of Ep,c (black circles) and ν(FeO) values (blue squares) versus E1/2(IV/III) vs. Fc+/0 for the FeIVO complexes in this study (data from Tables 2 and 1, respectively). The red line represents the best linear fit for the Ep,cvs. E1/2(IV/III) correlation with the red dot indicating an estimation of the E1/2(IV/III) value for 4 from the linear correlation. | ||
The plot shown in Fig. 4 emphasizes that the nature of the pentadentate ligand can have a profound effect on the redox potential of the FeIVO unit. Despite the fact that all five ligands are neutral in charge and consist only of combinations of tertiary amine and pyridine donors, there is a difference of 0.3–0.4 V in the potentials for the complexes in the series. The main difference between the three complexes at the bottom half of this range (i.e.1, 3, and 4) and the two at the top (i.e.2 and 5) is the presence of at least one pyridine donor whose plane is oriented perpendicular to the FeO unit. In fact 5 has two such pyridine donors perpendicular to the FeO unit and exhibits the highest potential among the complexes in the series. In contrast, 4, which is an isomer of 5 with no perpendicular pyridine rings, has a considerably lower potential that is comparable to those of 1 and 3. In an earlier DFT study of the possible isomers of 2, we noted the greater instability of isomers with a larger number of perpendicular pyridine donors,36 a notion supported by the experimental observations on 5. The difference in stability has been attributed to steric effects of the α-H on the perpendicular pyridine donor, resulting in a longer calculated Fe–N bond distance. Also shown in Fig. 4 is a plot of the ν(FeO) values observed for the five complexes by resonance Raman spectroscopy relative to their E1/2(IV/III) values. Interestingly, 2 and 5, the complexes with the higher potentials exhibit lower ν(FeO) frequencies that presumably reflect weaker, and perhaps more reactive, FeO bonds engendered by the interactions of the pentadentate ligands with the FeIVO unit. This study provides for the first time the E1/2(IV/III) potentials of five related oxoiron(IV) complexes directly determined by electrochemical methods. Complexes 1–5 thus represent an interesting series with which to probe the oxidative reactivity of the FeIVO unit.
C. Oxidative reactivity
Table 3 compares kinetic data for the five complexes with respect to their rates of oxo-atom transfer and H-atom abstraction. In one set of experiments, the oxidation of thioanisole by 1–5 was investigated in CH3CN solutions at −10 °C, resulting in about a 90% yield of methyl phenyl sulfoxide (Table 3). The progress of oxo-transfer could be monitored by the loss of the characteristic near-IR bands of the oxoiron(IV) complexes. For 1 and 3, formation 1a and 3a could also be conveniently monitored by the appearance of their characteristic intense visible chromophores at 458 nm (ε ∼ 4000 M−1 cm−1)69 and 455 nm (ε ∼ 2600 M−1 cm−1), respectively. Thus 1–5 are quite effective at oxo-transfer to thioanisole. Pseudo-first-order kinetic behavior was observed over the entire course of the reactions, and second order rate constants were extracted from the dependence of the pseudo-first-order rate constants on thioanisole concentration (Table 3, Fig. S5†). From these results, a reactivity order of 5 > 2 > 4 > 1 > 3 was obtained. Furthermore, when the logarithms of the second order rate constants were plotted versus the E1/2(IV/III) values obtained from spectropotentiometric experiments in CH3CN containing 0.1 M H2O, an excellent linear correlation was observed (Fig. 5), demonstrating that the FeIV/III potentials reflect the relative electrophilicities of the FeO units in these complexes.
| Complex | 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|---|
| E 1/2(IV/III) vs. Fc+/0 in CH3CN with 0.1 M water | 0.90 | 1.07 | 0.83 | (0.98)a | 1.23 | |
| a Value estimated from the correlation in Fig. 4. b From ref. 47. c See Fig. S9–S12.1 d See Fig. S12 and S13.1 | ||||||
| k 2,c M−1 s−1 (% yield of oxidation product) [KIE]d | CHD (−40 °C) | 0.070(3) (89%) | 0.96(3) (99%) | 0.027(1) (92%) | 0.014(1) (88%) | 0.37(2) (89%) |
| DHA (25 °C) | 18(1) [20]b | 100(6) [16] | 7.4(1) [31] | 1.1(1) [13] | 40(3) [12] | |
| PhCH2OH (25 °C) | 0.10(1) (80%) | 0.86(2) (83%) | 0.042(1) (80%) | 0.013(1) (85%) | 0.33(2) (90%) | |
| PhSMe (−10 °C) | 0.014 (90%) | 0.33 (93%) | 0.004 (90%) | 0.024 (75%) | 2.4 (95%) | |
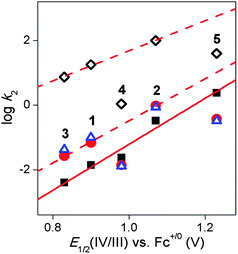 | ||
| Fig. 5 Plots of the logarithms of the second-order rate constants for the oxidation of thioanisole at −10 °C (filled black squares), CHD at −40 °C (filled red circles), PhCH2OH at 25 °C (open blue triangles) and DHA at 25 °C (open black diamonds) in CH3CN vs. the E1/2(IV/III) value measured by spectropotentiometry for oxoiron(IV) complexes 1–5. The red solid line is defined by the rates of thioanisole oxidation by 1–5, while the red dashed lines are defined by the rates of H-atom abstraction from various substrates by 1–3. The red dashed lines show that the H-atom abstraction rates associated with 4 and 5 are about an order of magnitude lower than predicted. | ||
The thioanisole oxidation results of the five ferryl complexes were further corroborated by a study of the oxo-transfer abilities of 1–5 to the various iron(II) precursors 1a–5a at 25 °C. Previously Nam and co-workers42 established a reactivity order of 2 > 1 > [FeIV(O)(TMC)(NCCH3)]2+ in their investigation of the reactions of an oxoiron(IV) complex of one ligand and the iron(II) complex of a different ligand.42 The present study extends the series to include 3, 4, and 5. Thus, addition of 3a (1.5 mM) to an equimolar solution of 1 in CH3CN resulted in the disappearance of the 695 nm band of 1 and the concomitant quantitative formation of the near-IR features of 3 (Fig. S6†), indicating that 1 is more oxidizing than 3. The other oxoiron(IV) complexes, 2, 4, and 5, were also found to convert 3a to 3 quantitatively, while the addition of 1a, 2a, 4a, or 5a to 3 did not result in the respective formation of 1, 2, 4, or 5. On the other hand, 5 was found to transfer its oxo atom to 1a, 2a, 3a, and 4a. Because of the similarity of the near IR features of 2 and 5, the reaction between 5 and 2a was monitored instead by ESI-MS, which is shown in Fig. S7.† Mixing equimolar amounts of 2a and 5 resulted in a decrease in the signals corresponding to [FeII(BnTPEN)(CF3SO3)]+ (m/z = 628.12) and {[FeIV(O)(BP2)](CF3SO3)}+ (m/z = 736.01) with a concomitant increase in the signals corresponding to {[FeIV(O)(BnTPEN)](CF3SO3)}+ (m/z = 644.04) and [FeII(BP2)(CF3SO3)]+ (m/z = 720.11). In contrast, no oxo-transfer was observed in the reverse reaction between 2 and 5a. The oxo-transfer ability of 4 was found to be significantly lower than that of its structural isomer 5, 4 being able to transfer its oxo group only to 1a (Fig. S8†) and 3a, but not to 2a. So 3a is the universal oxo-atom acceptor in the series, while 5 is the most effective oxo-transfer donor. The intermetal oxo-transfer experiments thus confirm the reactivity order of 5 > 2 > 4 > 1 > 3 that was observed for the oxidation of thioanisole. Therefore the relative OAT reactivities of the FeIV(O) complexes appear to be independent of the nature of the oxygen atom acceptor (i.e. thioanisole and [FeII(LN5)]2+), despite their having fundamentally distinct chemical structures.
For comparison, the hydrogen atom abstraction rates of 1–5 were also investigated with three substrates: 1,3-cyclohexadiene (CHD), 9,10-dihydroanthracene (DHA), and benzyl alcohol (Fig. S9–S13†). An earlier study of 1 and 2 found linear correlations in plots of the logarithms of the second order oxidation rate constants (normalized on a per hydrogen basis) for a large number of hydrocarbon substrates versus their DC–H values (81–99 kcal mol−1),39,47 consistent with C–H bond cleavage by the FeIVO unit as the rate determining step. In addition, large kinetic isotope effects (KIE) were found for the cleavage of C–H bonds of a number of hydrocarbon substrates. For this study, large KIE values for the oxidation of DHA were observed for all five complexes (Table 3, Fig. S12 and S13†), demonstrating that C–H bond cleavage is a significant component of the rate determining step. Furthermore all KIE values were greater than the semi-classical limit of 7 (Table 3), suggesting that hydrogen tunnelling likely plays a role in the H-atom abstraction step.
Interestingly, the linear correlation observed for the log k2 values for thioanisole oxidation versus E1/2(IV/III) values does not extend to rates for C–H bond cleavage by 1–5 (Fig. 5, Table 3). While the respective log k′2 values associated with 1–3 for the oxidation of CHD, DHA, and benzyl alcohol respectively fall on lines that are approximately parallel to the line obtained for thioanisole oxidation by all five complexes, the bispidine complexes 4 and 5 deviate from this pattern and exhibit H-atom abstraction rates that are about an order of magnitude below what are predicted by these lines. Clearly, the oxo-transfer and H-atom abstraction reactions are not governed by a common set of factors.
Our observations can be compared with the results of two previous studies. In one investigation, Fukuzumi and co-workers studied the dependence of rate constants in OAT and HAT reactions of cumylperoxyl radical versus the redox potentials of the substrates.70 The general trend observed for reactions with phosphines and sulfides showed an increase in OAT rates with a decrease in the oxidation potential of the substrate. HAT rates from N,N-dimethylanilines show a similar trend, but they plateaued with the introduction of electron donating substituents, suggesting some mechanistic complexity. Although the Fukuzumi study focused on the effects of substrate redox potential, and not of the oxidant, the conclusions reached may be regarded as coincident with ours, showing that reaction rates generally correlate with the electrochemical driving force of the reaction. In another study, Sastri et al. correlated redox potentials with OAT and HAT reaction rates of a series of [FeIV(O)(TMC)(X)] complexes (X = NCCH3, −O2CCF3, −N3, or −SR).38 It was found that the OAT rates increased with the electrophilicity of the oxoiron(IV) center, as reflected by the FeIV/III potential observed by cyclic voltammetry, which decreased as the axial ligand became more basic. In contrast, the HAT rates for the same series showed the opposite trend, where reaction rates increased with the basicity of the axial ligand. Clearly the trends observed in the present study do not follow those found in the TMC study. While both sets of OAT rates increase with higher FeIV/III potential, the HAT rates have opposite trends. It should be noted that the TMC series entails a systematic change of only the ligand trans to the oxo group, while the present study involves changes in the ligands cis to the oxo group. These two sets of data suggest that the factors governing the rate of H-atom abstraction are likely to be complex and demand further scrutiny.
Unlike oxo-transfer, which is a 2-e− oxidation process, H-atom abstraction generally involves rate determining transfer of one electron coupled with a proton. Applying the Bordwell–Polanyi relationship to metal–oxo complexes, Mayer71–73 has argued that the rate of H-atom abstraction by metal–oxo complexes correlates linearly with the strength of the MO–H bond (DO–H) that is formed. The DO–H value in turn is determined by the 1-e− Mn+/(n−1)+ (M = transition metal ion) redox potential and the pKa of the nascent O–H bond; thus the latter is an additional factor that can be modulated by the nature of the pentadentate supporting ligands, which vary in structure and in the number of amine and pyridine donors. Unfortunately, the pKa values of the FeIIIO–H species are not straightforward to measure. On the assumption that Mayer's approach applies for the five complexes in this study, the lower than expected C–H bond cleaving reactivity of 4 and 5 may result from their corresponding FeIIIO–H species having pKa's that are lower than those of 1–3.
Alternatively, the deviation of the C–H bond cleaving rates from what is expected by considering only the electrophilicity of the oxoiron(IV) complex may arise from differences in electronic structure engendered by the different pentadentate ligands. According to the two-state reactivity (TSR) concept developed by Shaik,74–78 the rate of H-atom abstraction by an S = 1 FeIVO complex is governed by a more reactive low lying S = 2 excited state that becomes populated as the reaction traverses along the reaction coordinate. DFT calculations on the [FeIV(O)(TMC)(X)] series showed that the energy gap between the S = 1 ground state and the S = 2 excited state decreased with increasing basicity of the axial ligand, thereby making the least electrophilic complex in the series the most reactive for H-atom abstraction.78 To determine whether the TSR rationale can explain the reactivity behavior of 1–5 requires a measurement of the zero field splitting (ZFS) of the S = 1 ground states of the five complexes. The zero field splitting reflects the extent of mixing between the triplet ground state and the quintet excited state via spin-orbit coupling, which can be estimated by determining the zero field splitting parameter D of each complex by high-field Mössbauer spectroscopy (with an uncertainly of ±2–3 cm−1) or, with higher precision, by high frequency EPR spectroscopy (±0.05 cm−1).79 Carrying out such measurements on these complexes would clearly be helpful.
Stereoelectronic effects may also play a role in modulating the H-atom abstraction rates of 1–5. From a consideration of the frontier molecular orbitals (FMOs) of the S = 1 oxoiron(IV) unit, it has been postulated that the attack of the substrate C–H bond by the oxoiron(IV) unit mainly involves the interaction between the π* MOs of the oxoiron(IV) unit and the target substrate C–H bond, requiring an FeO–HC angle of 120°.56,80,81 On the other hand, consideration of the FMOs of the S = 2 oxoiron(IV) unit suggests that C–H bond attack can also involve the σ* MO that would ideally require an FeO–HC angle of 180°. Based on a Nuclear Resonance Vibrational Spectroscopy (NRVS) study of 1,82 Solomon postulated that the α-H-atoms on the pyridine ligands present a barrier for the π-approach of substrate C–H bonds towards the oxoiron(IV) unit that would decrease the rate of H-atom abstraction. This notion was supported by the observed higher reactivity of an S = 1 oxoiron(IV) complex related to 1, where two of the pyridine donors were replaced with less bulky carboxylates.83 While similar considerations should apply to the other complexes, such stereoelectronic factors may also be influenced by ligand topology, a variety of which is represented by the pentadentate ligands that support the FeO units in 1–5 (Scheme 1). In addition, the greater rigidity of the bicyclic bispidine ligand framework may impose additional constraints to the reactivity of the FeO unit that lead to the lower than expected reactivity of 4 and 5. Particularly germane to the above discussion are two papers by Nam and co-workers. In one paper was compared the oxidative reactivity of the topological isomers of [FeIV(O)(BQCN)(NCCH3)]2+ (BQCN = N,N′-dimethyl-N,N′-bis(8-quinolyl)cyclohexane-1,2-diamine) where the cis-α isomer was found to be much more reactive than the cis-β isomer.84 In another paper a comparison of [FeIV(O)(TMC)(NCCH3)]2+ and [FeIV(O)(TBC)(NCCH3)]2+ (TBC = 1,4,8,11-tetrabenzylcyclam) was reported,22 where the oxidative properties of the relatively unreactive TMC complex were found to be enhanced 100-fold by the substitution of the methyl groups by benzyl groups, demonstrating that simple substitutions can have profound effects on reactivity.
In the above discussion of HAT reactivity, an implicit assumption has been made that hydrogen-atom abstraction occurs by the same mechanism for all five complexes. While this assumption is plausible, it may not necessarily be the case for these five complexes. Besides redox potential, additional considerations include the electron transfer rate (reorganization energies) and the question of whether these reactions are truly concerted or instead are dominated by electron transfer or proton transfer. In fact, the observation that 4 and 5 have lower KIEs for DHA oxidation than the other three complexes (Table 3) may suggest a HAT mechanism for the two bispidine complexes that is somewhat different from that of the other three complexes. Clearly, more studies are needed to clarify the mechanistic picture.
In summary, we have characterized the structural and spectroscopic properties of five synthetic S = 1 oxoiron(IV) complexes 1–5 supported by pentadentate ligands with combinations of pyridine and amine donors. We have also measured their FeIV/III redox potentials by spectropotentiometry, providing for the first time electrochemical determination of the E1/2(IV/III) redox potentials for a series of oxoiron(IV) complexes. We have compared their redox properties with respect to their abilities to carry out oxo-atom transfer and H-atom abstraction. While the measured oxo-transfer rates to thioanisole nicely correlate with redox potential, this is not the case for H-atom abstraction, demonstrating that the latter transformation is governed by factors more complex than for oxo-atom transfer.
Acknowledgements
This work was supported by grants from the US National Institutes of Health (GM-33162 to LQ and postdoctoral fellowship GM-75700 to TAJ), the US National Science Foundation (CHE1058248 to LQ), the German Science Foundation (DFG to PC), MCYT of Spain, (CTQ2009-08464/BQU to MC and PhD grant to LG) and European Research Council (ERC-29910 to MC), and Generalitat de Catalunya (Icrea Acadèmia to MC and 2009SGR637). We thank Dr Yuming Zhou for his assistance in obtaining stopped flow kinetic data for the oxidation of DHA at room temperature. XAS data were collected on beamlines X9B and X3B at the National Synchrotron Light Source (NSLS), Brookhaven National Laboratory. NSLS is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract no. DE-AC02-98CH10886. NSLS beamlines X9B and X3B were supported by the National Institute for Biomedical Imaging and Bioengineering under P41-EB-001979. We thank Dr Alexander Ignatiev and Dr Sandeep Rekhi for technical assistance with our XAS experiments.References
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–349 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS.
- M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS.
- J. C. Price, E. W. Barr, T. E. Glass, C. Krebs and J. M. Bollinger, Jr, J. Am. Chem. Soc., 2003, 125, 13008–13009 CrossRef CAS.
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger, Jr and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef CAS.
- P. J. Riggs-Gelasco, J. C. Price, R. B. Guyer, J. H. Brehm, E. W. Barr, J. M. Bollinger, Jr and C. Krebs, J. Am. Chem. Soc., 2004, 126, 8108–8109 CrossRef CAS.
- D. A. Proshlyakov, T. F. Henshaw, G. R. Monterosso, M. J. Ryle and R. P. Hausinger, J. Am. Chem. Soc., 2004, 126, 1022–1023 CrossRef CAS.
- L. M. Hoffart, E. W. Barr, R. B. Guyer, J. M. Bollinger, Jr and C. Krebs, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14738–14743 CrossRef CAS.
- D. P. Galonic, E. W. Barr, C. T. Walsh, J. M. Bollinger, Jr and C. Krebs, Nat. Chem. Biol., 2007, 3, 113–116 CrossRef CAS.
- M. L. Matthews, C. M. Krest, E. W. Barr, F. H. Vaillancourt, C. T. Walsh, M. T. Green, C. Krebs and J. M. Bollinger, Jr, Biochemistry, 2009, 48, 4331–4343 CrossRef CAS.
- A. J. Panay, M. Lee, C. Krebs, J. M. Bollinger, Jr and P. F. Fitzpatrick, Biochemistry, 2011, 50, 1928–1933 CrossRef CAS.
- B. E. Eser, E. W. Barr, P. A. Frantom, L. Saleh, J. M. Bollinger, Jr, C. Krebs and P. F. Fitzpatrick, J. Am. Chem. Soc., 2007, 129, 11334–11335 CrossRef CAS.
- D. G. Fujimori, E. W. Barr, M. L. Matthews, G. M. K. J. R. Yonce, C. T. Walsh, J. M. Bollinger, Jr, C. Krebs and P. J. Riggs-Gelasco, J. Am. Chem. Soc., 2007, 129, 13408–13409 CrossRef CAS.
- L. Que, Jr, Acc. Chem. Res., 2007, 40, 493–500 CrossRef.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS.
- J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef.
- A. R. McDonald and L. Que, Jr, Coord. Chem. Rev., 2012 DOI:10.1016/j.ccr.2012.1008.1002.
- C. A. Grapperhaus, B. Mienert, E. Bill, T. Weyhermuller and K. Wieghardt, Inorg. Chem., 2000, 39, 5306–5317 CrossRef CAS.
- J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que, Jr, Science, 2003, 299, 1037–1039 CrossRef CAS.
- M. R. Bukowski, K. D. Koehntop, A. Stubna, E. L. Bominaar, J. A. Halfen, E. Münck, W. Nam and L. Que, Jr, Science, 2005, 310, 1000–1002 CrossRef CAS.
- T. A. Jackson, J. U. Rohde, M. S. Seo, C. V. Sastri, R. De Hont, A. Stubna, T. Ohta, T. Kitagawa, E. Münck, W. Nam and L. Que, Jr, J. Am. Chem. Soc., 2008, 130, 12394–12407 CrossRef CAS.
- S. A. Wilson, J. Chen, S. Hong, Y.-M. Lee, M. Clémancey, R. García-Serres, T. Nomura, T. Ogura, J.-M. Latour, B. Hedman, K. O. Hodgson, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2012, 134, 11791–11806 CrossRef CAS.
- M. R. Bukowski, P. Comba, A. Lienke, C. Limberg, C. L. d. Laorden, R. Mas-Balleste, M. Merz and L. Que, Jr, Angew. Chem., Int. Ed., 2006, 45, 3446–3449 CrossRef CAS.
- J. Bautz, M. R. Bukowski, M. Kerscher, A. Stubna, P. Comba, A. Lienke, E. Münck and L. Que, Jr, Angew. Chem., Int. Ed., 2006, 45, 5681–5684 CrossRef CAS.
- P. Comba, S. Fukuzumi, H. Kotani and S. Wunderlich, Angew. Chem., Int. Ed., 2010, 49, 2622–2625 CrossRef CAS.
- P. Comba, M. Kerscher and W. Schiek, Prog. Inorg. Chem., 2008, 55, 613–704 CrossRef.
- M. H. Lim, J.-U. Rohde, A. Stubna, M. R. Bukowski, M. Costas, R. Y. N. Ho, E. Münck, W. Nam and L. Que, Jr, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3665–3670 CrossRef CAS.
- A. Company, I. Prat, J. R. Frisch, R. Mas-Ballest, M. Guell, G. Juhasz, X. Ribas, E. Münck, J. M. Luis, L. Que, Jr and M. Costas, Chem.–Eur. J., 2011, 17, 1622–1634 CrossRef CAS.
- M. S. Seo, N. H. Kim, K.-B. Cho, J. E. So, S. K. Park, M. Clemancey, R. Garcia-Serres, J.-M. Latour, S. Shaik and W. Nam, Chem. Sci., 2011, 2, 1039–1045 RSC.
- O. Pestovsky, S. Stoian, E. L. Bominaar, X. Shan, E. Münck, L. Que, Jr and A. Bakac, Angew. Chem., Int. Ed., 2005, 44, 6871–6874 CrossRef CAS.
- J. England, M. Martinho, E. R. Farquhar, J. R. Frisch, E. L. Bominaar, E. Münck and L. Que, Jr, Angew. Chem., Int. Ed., 2009, 48, 3622–3626 CrossRef CAS.
- D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2010, 132, 12188–12190 CrossRef CAS.
- J. England, Y. Guo, K. M. V. Heuvelen, M. A. Cranswick, G. T. Rohde, E. L. Bominaar, E. Münck and L. Que, Jr, J. Am. Chem. Soc., 2011, 133, 11880–11883 CrossRef CAS.
- J. P. Bigi, W. H. Harman, B. Lassalle-Kaiser, D. M. Robles, T. A. Stich, J. Yano, R. D. Britt and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 1536–1542 CrossRef CAS.
- A. Thibon, J. England, M. Martinho, V. G. Young Jr, J. R. Frisch, R. Guillot, J.-J. Girerd, E. Münck, L. Que, Jr and F. Banse, Angew. Chem., Int. Ed., 2008, 47, 7064–7067 CrossRef CAS.
- E. J. Klinker, J. Kaizer, W. W. Brennessel, N. L. Woodrum, C. J. Cramer and L. Que, Jr, Angew. Chem., Int. Ed., 2005, 44, 3690–3694 CrossRef CAS.
- J. England, Y. Guo, E. R. Farquhar, V. G. Young Jr, E. Münck and L. Que, Jr, J. Am. Chem. Soc., 2010, 132, 8635–8644 CrossRef CAS.
- C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que, Jr, S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS.
- J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que, Jr, J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef CAS.
- P. Comba, M. Maurer and P. Vadivelu, J. Phys. Chem. A, 2008, 112, 13028–13036 CrossRef CAS.
- P. Comba, M. Maurer and P. Vadivelu, Inorg. Chem., 2009, 48, 10389–10396 CrossRef CAS.
- C. V. Sastri, K. Oh, Y. J. Lee, M. S. Seo, W. Shin and W. Nam, Angew. Chem., Int. Ed., 2006, 45, 3992–3995 CrossRef CAS.
- Y.-M. Lee, H. Kotani, T. Suenobu, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 434–435 CrossRef CAS.
- G. Roelfes, M. Lubben, K. Chen, R. Y. N. Ho, A. Meetsma, S. Genseberger, R. M. Hermant, R. Hage, S. K. Mandal, V. G. Young Jr, Y. Zang, H. Kooijman, A. L. Spek, L. Que, Jr and B. L. Feringa, Inorg. Chem., 1999, 38, 1929–1936 CrossRef CAS.
- G. Roelfes, V. Vrajmasu, K. Chen, R. Y. N. Ho, J.-U. Rohde, C. Zondervan, R. M. l. Crois, E. P. Schudde, M. Lutz, A. L. Spek, R. Hage, B. L. Feringa, E. Münck and L. Que, Jr, Inorg. Chem., 2003, 42, 2639–2653 CrossRef CAS.
- M. J. Collins, K. Ray and L. Que, Jr, Inorg. Chem., 2006, 45, 8009–8011 CrossRef CAS.
- E. J. Klinker, S. Shaik, H. Hirao and L. Que, Jr, Angew. Chem., Int. Ed., 2009, 48, 1291–1295 CrossRef CAS.
- R. Gupta and A. S. Borovik, J. Am. Chem. Soc., 2003, 125, 13234–13242 CrossRef CAS.
- B. A. Moyer and T. J. Meyer, Inorg. Chem., 1981, 20, 436–444 CrossRef CAS.
- K. J. Takeuchi, M. S. Thompson, D. W. Pipes and T. J. Meyer, Inorg. Chem., 1984, 23, 1845–1851 CrossRef CAS.
- R. C. McHatton and F. C. Anson, Inorg. Chem., 1984, 23, 3935–3942 CrossRef CAS.
- A. A. Diamantis, W. R. Murphy Jr and T. J. Meyer, Inorg. Chem., 1984, 23, 3230–3234 CrossRef CAS.
- M. E. Marmion and K. J. Takeuchi, J. Am. Chem. Soc., 1986, 108, 510–511 CrossRef CAS.
- M. E. Marmion and K. J. Takeuchi, J. Am. Chem. Soc., 1988, 110, 1472–1480 CrossRef CAS.
- A. Decker, J.-U. Rohde, L. Que, Jr and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 5378–5379 CrossRef CAS.
- A. Decker, J.-U. Rohde, E. J. Klinker, S. D. Wong, L. Que, Jr and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 15983–15996 CrossRef CAS.
- J.-U. Rohde, S. Torelli, X. Shan, M. H. Lim, E. J. Klinker, J. Kaizer, K. Chen, W. Nam and L. Que, Jr, J. Am. Chem. Soc., 2004, 126, 16750–16761 CrossRef CAS.
- J.-U. Rohde, A. Stubna, E. L. Bominaar, E. Münck, W. Nam and L. Que, Jr, Inorg. Chem., 2006, 45, 6435–6445 CrossRef CAS.
- A. E. Anastasi, P. Comba, J. McGrady, A. Lienke and H. Rohwer, Inorg. Chem., 2007, 46, 6420–6426 CrossRef CAS.
- D. Wang, M. Zhang, P. Buhlmann and L. Que, Jr, J. Am. Chem. Soc., 2010, 132, 7638–7644 CrossRef CAS.
- M. R. Egmond, P. M. Fasella, G. A. Veldink, J. F. G. Vliegenthart and J. Boldingh, Eur. J. Biochem., 1977, 76, 469–479 CrossRef CAS.
- R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 1997, 119, 8566–8567 CrossRef CAS.
- C. R. Goldsmith and T. D. P. Stack, Inorg. Chem., 2006, 45, 6048–6055 CrossRef CAS.
- J. Mukherjee, R. L. Lucas, M. K. Zart, D. R. Powell, V. W. Day and A. S. Borovik, Inorg. Chem., 2008, 47, 5780–5786 CrossRef CAS.
- N. Ortega-Villar, V. M. Ugalde-Saldívar, B. Flores-Pérez, M. Flores-Alamo, J. A. Real and R. Moreno-Esparza, Inorg. Chim. Acta, 2011, 375, 213–219 CrossRef CAS.
- J. Park, Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 3903–3911 CrossRef.
- A. Dovletoglou, S. A. Adeyemi and T. J. Meyer, Inorg. Chem., 1996, 35, 4120–4127 CrossRef CAS.
- Y. Hirai, T. K. Y. Mizutani, Y. Shiota, K. Yoshizawa and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 5772–5776 CrossRef CAS.
- M. Lubben, A. Meetsma, E. C. Wilkinson, B. Feringa and L. Que, Jr, Angew. Chem., Int. Ed. Engl., 1995, 34, 1512–1514 CrossRef CAS.
- S. Fukuzumi, K. Shimoosako, T. Suenobu and Y. Watanabe, J. Am. Chem. Soc., 2003, 125, 9074–9082 CrossRef CAS.
- J. M. Mayer, Acc. Chem. Res., 1998, 31, 441–450 CrossRef CAS.
- J. R. Bryant and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 10351–10361 CrossRef CAS.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS.
- S. Shaik, D. Danovich, A. Fiedler, D. Schroder and H. Schwarz, Helv. Chim. Acta, 1995, 78, 1393–1407 CrossRef CAS.
- D. Schroder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139–145 CrossRef CAS.
- H. Hirao, D. Kumar, L. Que, Jr and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8590–8606 CrossRef CAS.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS.
- H. Hirao, L. Que, Jr, W. Nam and S. Shaik, Chem.–Eur. J., 2008, 14, 1740–1756 CrossRef CAS.
- J. Krzystek, J. England, K. Ray, A. Ozarowski, D. Smirnov, L. Que, Jr and J. Telser, Inorg. Chem., 2008, 47, 3483–3485 CrossRef CAS.
- C. Geng, S. Ye and F. Neese, Angew. Chem., Int. Ed., 2010, 49, 5717–5720 CrossRef CAS.
- S. Shaik, H. Chen and D. Janardanan, Nat. Chem., 2010, 3, 19–27 CrossRef.
- S. D. Wong, C. B. Bell, III, L. V. Liu, Y. Kwak, J. England, E. E. Alp, J. Zhao, L. Que, Jr and E. I. Solomon, Angew. Chem., Int. Ed., 2011, 50, 3215–3218 CrossRef CAS.
- A. R. McDonald, Y. Guo, V. V. Vu, E. L. Bominaar, E. Münck and L. Que, Jr, Chem. Sci., 2012, 3, 1680–1693 RSC.
- S. Hong, Y.-M. Lee, K.-B. Cho, K. Sundaravel, J. Cho, M. J. Kim, W. Shin and W. Nam, J. Am. Chem. Soc., 2011, 133, 11876–11879 CrossRef CAS.
- A. Draksharapu, Q. Li, H. Logtenberg, T. A. van den Berg, A. Meetsma, J. S. Killeen, B. L. Feringa, R. Hage, G. Roelfes and W. R. Browne, Inorg. Chem., 2012, 51, 900–913 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, experimentally observed and calculated ESI-MS spectra for 3, additional cyclic voltammetry traces for 1a–5a, EPR and ESI-MS data for 1b–5b, additional details of inter-complex oxo-atom transfer reactions, plots showing the determination of pseudo first order and second order rate constants for reactions of 1–5 and the KIE values, and details of the EXAFS analyses. See DOI: 10.1039/c2sc21318d |
| ‡ Current address: Humboldt-Universität zu Berlin, Department of Chemistry, Brook-Taylor Str. 2, 12489 Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2013 |