Aggregate morphology of nano-TiO2: role of primary particle size, solution chemistry, and organic matter
Indranil
Chowdhury
a,
Sharon L.
Walker
b and
Steven E.
Mylon
*c
aNational Exposure Research Laboratory, United States Environmental Protection Agency, Athens, GA, USA. E-mail: Chowdhury.Indranil@epa.gov; Tel: +1 706-355-8341
bDepartment of Chemical and Environmental Engineering, University of California, Riverside, CA, USA. E-mail: swalker@engr.ucr.edu; Fax: +1 951-827-5696; Tel: +1 951-827-6094
cDepartment of Chemistry, Lafayette College, Easton, PA, USA. E-mail: mylons@lafayette.edu; Fax: +1 610-330-5714; Tel: +1 610-330-5825
First published on 28th November 2012
Abstract
A systematic investigation was conducted to understand the role of aquatic conditions on the aggregate morphology of nano-TiO2, and the subsequent impact on their fate in the environment. In this study, three distinctly sized TiO2nanoparticles (6, 13, and 23 nm) that had been synthesized with flame spray pyrolysis were employed. Nanoparticle aggregate morphology was measured using static light scattering (SLS) over a wide range of solution chemistry, and in the presence of natural organic matter (NOM). Results showed that primary nanoparticle size can significantly affect the fractal dimension of stable aggregates. A linear relationship was observed between surface areas of primary nanoparticles and fractal dimension indicating that smaller primary nanoparticles can form more compact aggregate in the aquatic environment. The pH, ionic strength, and ion valence also influenced the aggregate morphology of TNPs. Increased pH resulted a decrease in fractal dimension, whereas higher ionic strength resulted increased fractal dimension particularly for monovalent ions. When NOM was present, aggregate fractal dimension was also affected, which was also notably dependent on solution chemistry. Fractal dimension of aggregate increase for 6 nm system in the presence of NOM, whereas a drop in fractal dimension was observed for 13 nm and 23 nm aggregates. This effect was most profound for aggregates comprised of the smallest primary particles suggesting that interactions of NOM with smaller primary nanoparticles are more significant than those with larger ones. The findings from this study will be helpful for the prediction of nanoparticle aggregate fate in the aquatic environment.
Environmental impactThe fate, transport and bioavailability of nanoparticles in aquatic systems are linked to their aggregation state. In this work, the morphology of nanoparticle aggregates (represented as fractal dimension) has been correlated with primary particle size and aquatic chemistry. Both solution chemistry, and presence of natural organic matter can also notably affect the morphology of nanoparticle aggregates. For the transport of nanoparticles aggregates in saturated systems, an earlier study revealed that the lower fractal dimensions resulted in higher break up potential during the transport through porous media. Thus a keen understanding of how solution conditions affect aggregate morphology is necessary for future prediction of nanomaterial aggregation states in the aquatic environment. |
1. Introduction
Anthropogenic nanomaterials are generating scientific interest due to their small size, and surface structure, which can lead to different properties than their bulk counterparts.1 Nanotechnology is one of the fastest growing technologies in the current century, which is predicted to be trillion dollar industry by 2015.2,3 However, with the increased production of nanomaterials, it is reasonable to expect their release in the environment creating unknown consequences in future. Hence, understanding the fate, and transport of engineered nanomaterials is essential for the environmental sustainability of nanotechnology. Though recent interest is primarily focused on potential toxicity of engineered nanomaterials, quite a few studies have been done on the fate and transport of these anthropogenic materials in the aquatic environment.1,4–13 Though most of the studies on transport primarily focused on the primary nanoparticles, a release of nanomaterials to a natural system may result in their formation into aggregates. Very few studies have so far focused on the transport of nanoparticle aggregates, and even fewer on the actual morphology of nanomaterial aggregates. This paper seeks to extend the knowledge on aggregate morphology of anthropogenic nanomaterials, particularly titanium dioxide in details, which will be helpful for prediction of their fate in the aquatic environment.Titanium dioxide nanoparticle (TNP) is one of the widely used nanomaterials with an approximate production of 4 million tons.14,15 Due to its unique UV absorbance and photocatalytic properties, TiO2 has been employed in consumer products, sunscreens, photocatalyst, and numerous other applications.3,15–17 Among different anthropogenic nanomaterials, it has been predicted that TiO2 will be found in the highest concentration in the aquatic environment.14,16 Hence, we chose TiO2 as model nanomaterials in this study.
Transport of TNPs has been found to be dependent on both physicochemical properties of nanomaterials as well as the aquatic chemistry.4,6,7,18–22 One of the most important factors affecting the transport, particularly for metal oxide nanomaterials, is aggregation.23–25 Aggregation of titanium dioxide nanomaterials has been shown to limit the mobility of these nanomaterials through porous media.23,24 Chowdhury et al.23 found that transport of TNPs through porous media is primarily governed by the aggregation state of nanomaterials, with larger aggregates straining out through packed-bed column. Break up of aggregates increased the elution of TNPs through porous media at high flow rates. Natural organic matter (NOM) also affected transport of TNP aggregates by reducing deposition of TNPs through porous media.10,26,27 Both electrostatic and electrosteric repulsion due to sorption of NOM reduced TNP aggregate size, which also assisted in transport through porous media. Microorganisms such as bacteria have also been reported to influence the aggregation state of TNPs affecting their transport as well.10,28
Morphology of particle aggregates is important for predicting transport behavior of nanomaterials in both natural and engineered systems, and this morphology differs depending on aquatic conditions such as pH and ionic strength. In the engineered systems such as a waste water treatment plant, not only solution chemistry, but also other parameters including microbes, organic matter, and treatment process can affect the morphology of aggregate which in turn affects their transport.29 Recent studies showed that break up of aggregate can enhance the transport of nano-TiO2 through porous media.29 Another study revealed that aggregate morphology can notably influence hydroxyl radical production of TNPs under UV light, which can consequently affect the toxicity.30
Though extensive research has been conducted on the aggregation of nanoparticles, there are very few studies on the morphology of the nanoparticle aggregates. Specifically, so far no study has been conducted how the primary particle size, solution chemistry, and presence of natural organic matter will affect the aggregate morphology. Since in the both natural and engineered systems, aquatic chemistry may differ considerably from time to time or from system to system, understanding the influence of these factors on the aggregate morphology of nanomaterials is essential for predicting the fate of nanomaterials in the environment. In this study, we investigated the role of primary particle size on the aggregate morphology of nano-TiO2, and effects of solution chemistry on this morphology. We found a useful correlation with the surface areas of primary nanoparticles and morphology, which may be employed for predicting aggregate morphology based on primary particle size. We also observed a notable influence of aquatic chemistry including pH, IS, ion valence, and presence of NOM on the formed aggregate.
2. Materials and methods
2.1 Synthesis, and characterization of distinctly sized TNPs
The detailed synthesis procedure and physical characterization of differently sized TNPs was described elsewhere.30 In brief, TNPs were produced using a flame spray pyrolysis technique31 from metallorganic precursor titanium(IV) isopropoxide (Strem Chemical, 99.9%). The primary particle size of TNPs was controlled at 6, 13 and 23 nm by adjusting the liquid precursor delivery rate and oxygen flow for atomization. The final nanoparticles formed by reaction and nucleation in the flame environment were collected from the glass filter. The primary particle size and crystal structure of the synthesized TNPs were determined from the X-ray diffraction measurements (Philips PW1800 diffractometer). The size of TNPs was further confirmed from transmission electron microscopy (TEM, FEI-PHILIPS CM300, Hillsboro, OR). Specific surface areas were also determined using nitrogen adsorption–desorption measurements (BET).2.2 Electrokinetic characterization of TNPs
Electrokinetic properties of distinctly sized TNPs were determined over a wide range of solution chemistry using a ZetaPALS analyzer (Brookhaven Instruments Corp., Holtsville, NY).10,23,25,32 pH was controlled at pH 7 and 10 in both monovalent (KCl) and divalent (CaCl2) ions over a range of 1 to 100 mM IS. Suwannee River Humic Acid (SRHA) standard II (International humic society substances, MN) was used as model organic matter. SRHA concentration was varied from 1 to 10 mg L−1. These ranges of solution chemistry, and NOM was chosen to simulate the conditions relevant to both natural and engineered aquatic systems.33 Analysis of variance (ANOVA) and student t-test was conducted to compare the difference among particles with 95% confidence level (P < 0.05).2.3 Aggregation kinetics of TNPs
Hydrodynamic diameter of TNPs were determined using dynamic light scattering (DLS) (Brookhaven Model BI-9000AT, Holtsville, NY) at a wavelength of 661 nm, and scattering angle of 90°. All solutions were made with ACS reagent grade chemicals (Fisher Scientific, Pittsburgh, PA), and pH was controlled with 10 mM HCl or KOH. For selected conditions, aggregation kinetics of TNPs were determined by DLS using ALV-CGS-3 (Langen, Germany) at a wavelength of 632.8 nm with 90° light scattering following the procedure mentioned elsewhere.19,34 Aggregation kinetics was determined in monovalent ions (KCl) for all sized TNPs, and data from these measurements was transformed to attachment efficiencies (α) using methods described previously.18,192.4 Determination of aggregate morphology
Static light scattering (SLS) technique was utilized to determine the morphology of TNP aggregates.35,36 A single-detector light scattering unit (ALV-CGS-3, Langen, Germany) was employed to make all SLS measurements.37 In ALV-CGS-3 compact goniometer system a 22 mW HeNe Laser (Uniphase) is employed to provide a single-frequency output with a wavelength of 632.8 nm. This light scattering system also has an ALV-proprietary optical fiber based detector, and an avalanche photodiode single photondetector. For light scattering measurements borosilicate culture tubes (Fisher Scientific, PA) were employed as sample cuvette. Scattering angle was varied from 30° to 140° to understand the influence of primary particle size on stable aggregate structure. Fractal dimension of TNP aggregates was determined from scattering intensities utilizing Rayleigh–Gans–Debye (RGD) theory from the following equations:| I(q) ∝ q−Df | (1) |
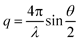 | (2) |
3. Results and discussion
3.1 Electrokinetic and hydrodynamic characterization of TNPs
The electrokinetic properties of TNPs are summarized in Table 1. In KCl, the EPMs of TNPs were highly negative at both pH 7 and 10 indicating that TNPs surfaces were negatively charged over the pH range of 7 to 10. This is consistent with a previous study showing that the isoelectric point of these TNPs was near pH 3.5.29 There was no significant difference observed in EPMs among different sized primary nanoparticles. Though EPMs remained quite similar for all three sets of TNPs, the aggregate size showed a notable dependence on primary particle size. Under almost all conditions, 6 nm primary particles formed stable aggregates of the largest size followed by 13 nm and 23 nm. A similar effect was also observed in a previous study that employed sets of differently sized hematite nanoparticles.38 As expected, an increase in IS (1 to 100 mM KCl) reduced EPMs due to charge screening at high salt condition for all TNPs.39 This electrical double layer compression at high IS resulted in more favorable aggregation conditions for all TNPs, and this is observed through larger aggregate sizes for each set of primary particles TNPs. An increase in pH from 7 to 10 resulted in a greater negative charge for all TNPs due to the hydroxylation in metal oxide nanomaterials.36 This increase in negative charge also reduced the size of TNP aggregates at pH 10.| 6 nm | 13 nm | 23 nm | |||||||
|---|---|---|---|---|---|---|---|---|---|
| EPMa | D agg b (nm) | D f c | EPM | D agg (nm) | D f | EPM | D agg (nm) | D f | |
| a Electrophoretic mobility. b Hydrodynamic diameter of aggregate (Dagg) was determined by dynamic light scattering. c Fractal dimension (Df) of aggregate was determined by static light scattering. d Suwannee River humic acid. | |||||||||
| pH 7 1 mM KCl | −2.60 ± 0.28 | 411.1 ± 58.8 | 2.07 | −2.61 ± 0.30 | 512.1 ± 26.9 | 1.78 ± 0.11 | −2.66 ± 0.26 | 442.3 ± 29.1 | 1.61 |
| pH 7 10 mM KCl | −1.54 ± 0.27 | 741.2 ± 76.7 | 2.08 | −1.66 ± 0.16 | 735.8 ± 86.7 | 1.88 ± 0.08 | −1.55 ± 0.19 | 717.1 ± 48.9 | 1.80 |
| pH 7 100 mM KCl | −0.87 ± 0.26 | 1399.86 ± 299.68 | 2.06 | −0.75 ± 0.29 | 1363.04 ± 264.34 | 1.92 | −0.80 ± 0.38 | 933.86 ± 271.67 | 1.86 |
| pH 7 10 mM CaCl2 | −0.38 ± 0.08 | 678.78 ± 70.03 | 1.91 | −0.53 ± 0.13 | 578.09 ± 98.81 | 1.84 ± 0.08 | −0.74 ± 0.13 | 589.48 ± 89.84 | 1.67 |
| pH 7 100 mM CaCl2 | 0.19 ± 0.63 | 1975.36 ± 517.83 | 2.32 | 0.35 ± 0.16 | 1901.94 ± 348.01 | 1.91 | −0.12 ± 0.50 | 1104.01 ± 188.91 | 1.70 |
| pH 10 1 mM KCl | −3.14 ± 0.34 | 181.5 ± 20.7 | 1.77 | −2.85 ± 0.14 | 145.5 ± 25.7 | 1.38 ± 0.05 | −3.35 ± 0.43 | 181.2 ± 6.0 | 1.05 |
| pH 10 10 mM KCl | −1.79 ± 0.26 | 414.2 ± 69.8 | 1.77 | −2.22 ± 0.28 | 455.1 ± 62.1 | 1.69 ± 0.14 | −2.04 ± 0.23 | 428.4 ± 30.0 | 1.88 |
| pH 10 10 mM CaCl2 | −0.65 ± 0.15 | 476.70 ± 42.59 | 1.78 | −0.40 ± 0.30 | 548.50 ± 87.63 | 1.94 | −0.84 ± 0.10 | 346.70 ± 35.68 | 1.78 |
| pH 7 10 mM KCl + SRHA 1 mg L−1 | −2.84 ± 0.19 | 204.12 ± 65.06 | 1.92 | −3.01 ± 0.25 | 219.54 ± 17.92 | 1.68 ± 0.03 | −3.41 ± 0.19 | 208.53 ± 7.40 | 1.31 |
| pH 7 100 mM KCl + SRHA 1 mg L−1d | −1.83 ± 0.24 | 1391.90 ± 254.30 | 1.77 | −2.23 ± 0.29 | 1331.32 ± 203.37 | 1.92 | −1.86 ± 0.24 | 712.88 ± 100.00 | 1.77 |
| pH 7 10 mM KCl + SRHA 10 mg L−1 | −3.19 ± 0.21 | 421.67 ± 90.56 | 2.38 | −3.34 ± 0.27 | 306.73 ± 26.89 | 1.69 | −3.36 ± 0.11 | 349.27 ± 30.0 | 1.14 |
| pH 7 10 mM CaCl2 + SRHA 1 mg L−1 | −0.81 ± 0.10 | 1439.92 ± 154.34 | 1.79 | −0.95 ± 0.06 | 988.08 ± 128.38 | 1.72 ± 0.04 | −0.99 ± 0.12 | 770.78 ± 79.64 | 1.65 |
| pH 7 100 mM CaCl2 + SRHA 1 mg L−1 | −0.15 ± 0.69 | 1607.99 ± 310.04 | 2.23 | −0.12 ± 0.65 | 1619.0 ± 226.44 | 1.81 | 0.02 ± 0.37 | 991.60 ± 79.13 | 1.74 |
| pH 10 10 mM KCl + SRHA 1 mg L−1 | −2.22 ± 0.12 | 276.37 ± 20.02 | 2.17 | −2.06 ± 0.27 | 272.77 ± 24.48 | 1.82 | −2.84 ± 0.31 | 222.71 ± 9.88 | 1.14 |
| pH 10 10 mM CaCl2 + SRHA 1 mg L−1 | −0.95 ± 0.08 | 1060.40 ± 138.34 | 2.25 | −0.99 ± 0.13 | 1104.01 ± 228.64 | 1.86 | −0.96 ± 0.15 | 621.03 ± 50.62 | 1.81 |
In divalent ions (CaCl2), the trends for electrokinetic and hydrodynamic properties of TNP aggregates remained similar to those observed in KCl. EPMs were significantly lower than those values in KCl due to effect of divalent ions.39 Similarly the aggregate size in CaCl2 was also significantly higher (700 to 2000 nm) than those in KCl.
In the presence of SRHA, TNPs became more negatively charged under all conditions. The increased negativity of metal oxide nanomaterials with addition of SRHA is due to the sorption of SRHA on TNP surfaces and the deprotonation of many acidic functionalities within the SRHA.10 In monovalent salts (KCl) the aggregate size also became notably smaller in the presence of SRHA both due to electrostatic and electrosteric repulsion.10 However, addition of SRHA in the presence of divalent ions (CaCl2) increased the TNP aggregate size. Previous studies showed that carboxylic functional groups of SRHA can bind with Ca2+ ions, and form large aggregates through bridging.10 There was significant influence of primary particle size observed even in the presence of SRHA. Though EPMs remained quite similar for TNP sizes, the hydrodynamic diameter for aggregates comprised of 6 nm primary nanoparticles remained the highest which is consistent with the trend observed in the absence of SRHA. As the SRHA concentration was increased from 1 to 10 mg L−1, we observed that the surface charges of the TNPs became slightly more negatively charged, oddly, however, aggregate size of TNPs actually increased with SRHA concentration. This may be due to higher concentration of SRHA leading to polymer bridging, which can increase the aggregate size.39
3.2 Aggregation of TNPs
In Fig. 1a, we present representative aggregation of differently sized TNPs as a function of time. Based on aggregation kinetics we constructed a plot of attachment efficiency of differently sized particles as a function of salt concentration in a similar fashion to some earlier papers.18,34,40 Based on attachment efficiencies we observe that 6 nm particles show more favorable attachment at similar conditions when compared to 13 nm and 23 nm. The critical coagulation concentration (CCC) of TNPs in monovalent ions (KCl) were found to be approximately 30 mM, 60 mM, and 100 mM for 6, 13, and 23 nm primary nanoparticles respectively. This is most likely due to the higher surface areas of smaller primary nanoparticles.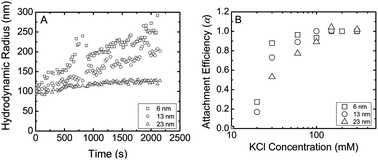 | ||
| Fig. 1 Aggregation kinetics of differently sized TiO2nanoparticles as a function of KCl concentration. (A) Aggregation kinetics in 30 mM KCl. (B) Attachment efficiency of TNPs as a function of KCl. TNP concentration was maintained at 1 mg L−1. Hydrodynamic radius was measured using dynamic light scattering. | ||
3.3 Aggregate morphology of TNPs
Upon the addition of SRHA we observed an important effect on the morphology of TNP aggregates. At 10 mM KCl, Df of aggregates decreased significantly with addition of SRHA, whereas at 100 mM KCl, the Df of 13 nm and 23 nm remained unchanged in the presence of SRHA. At lower IS sorption of SRHA on TNPs can reduce the compactness of aggregate because the adsorbed SRHA generally drives the CCC of the system to higher IS due to the addition of electrosteric repulsion on top of the electrostatic repulsion. However, as IS increased to 100 mM KCl, this subtle effect from addition of SRHA is diminished due to electrical double layer compression.39 The concentration of SRHA affected the Df as well. We observed a greater Df as we increased the concentration of SRHA from 1 to 10 mg L−1 in the 6 nm system but we observed a decrease in Df for 23 nm system, while Df remained unchanged for 13 nm particles. As a first approximation we suspect that the greater surface area of the 6 nm particles results in increased adsorption of SRHA, which can contribute to a higher degree of steric interactions between particles which decreases the attachment efficiencies of these particles. More compact aggregates are known to form under these conditions as particles have more opportunities to find the lowest energy configuration before attachment. For the 13 nm and 23 nm systems, there may be enough dissolved (unadsorbed) SRHA remaining in solution to result in more open aggregates.
At pH 7 and 10 mM CaCl2, the presence of SRHA resulted in a decrease in Df for 6 nm and 13 nm systems, whereas the 23 nm systems remained unchanged. However, Df increased with IS for all TNPs at 100 mM CaCl2 in the presence of SRHA. This is probably due to the saturation of most of the available divalent cation complexation sites within the SRHA. This neutralization of the SRHA functional groups may result in a collapse of the calcium saturated organic matter to the surface of the particle. These particles should now act like hard spheres that can pack tightly into aggregates. We observed that the dependence of Df on IS in this system was significantly higher for the 6 nm system compared with the 13 nm and 23 nm systems. At pH 10 in 10 mM KCl the addition of SRHA resulted a significant increase in Df for 6 nm and 13 nm-aggregates, whereas a drop in Df observed for 23 nm-aggregate. At pH 10 in 10 mM CaCl2 the addition of SRHA increased Df of the 6 nm system significantly, whereas there is a slight decrease in Df for 13 nm particles. However, Df remained virtually unchanged for 23 nm-aggregates. For many of these experimental conditions, the aggregates formed from the 6 nm primary particles possessed a high fractal dimension possibly due to rearrangement of these small dense particles to the lowest energy configuration – a more tightly packed aggregate. It is unlikely that we would observe further increases through changes in solution chemistry from this system. Overall, the fractal dimensions of TNP aggregates decreased with increases in primary particle size indicating that the primary particle size of those NPs that make up the aggregate plays an important role in driving aggregate morphology.
The observed effects of organic matter on the fractal dimensions of nanoparticle aggregates demonstrate the difficulty in understanding complex systems. As a first approximation the addition of organic matter might suggest the formation of more open aggregates, and hence result in lower fractal dimensions. The above results demonstrate otherwise probably due to the intricate nature of the physic-chemical interactions of the organic matter at the mineral water interface coupled with similar effects of solution chemistry. Solution chemistry including pH, ionic strength, ion valence, and presence of organic matter added additional complexity to this system of monodisperse nanoparticles, and because each variable can exert control of aggregate morphology in discreet ways teasing out the generalizations for similar particles becomes exceedingly difficult.
We do note that fractal dimension of TiO2 aggregates in this study did not follow the classic diffusion limited regime (Df ≈ 1.7) and reaction limited regime (Df ≈ 2.2) for few cases, particularly for 6 nm and 23 nm system in the presence of SRHA.39 For the 6 nm particles, Df was greater than 2.2 in the presence of SRHA. The increase of fractal dimension in the presence of natural organic matter has also been reported in other study.41 This is mainly due to increase in both electrostatic and electrosteric repulsive forces between particles in the presence of NOM, which can increase the collision opportunities between particles to form more compact aggregate. Aggregates can also restructure in the presence of SRHA, which can subsequently increase fractal dimension of formed aggregates. 23 nm system, on the other hand, resulted fractal dimension lower than 1.7, particularly in the presence of SRHA.
 | ||
| Fig. 2 Relative dependence of fractal dimension (Df) on surface area of primary particles as a function of (A) KCl only; (B) both KCl and SRHA; (C) CaCl2 only and (D) both CaCl2 and SRHA. | ||
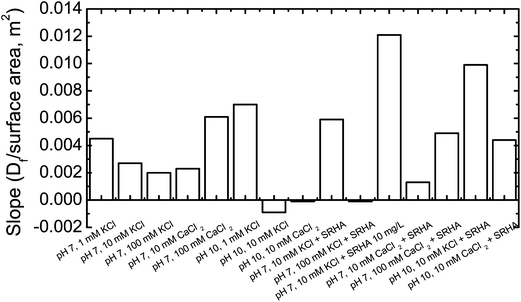 | ||
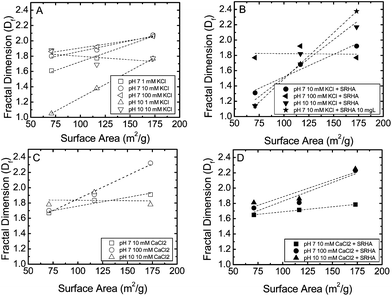
| Fig. 3 Sensitivity of fractal dimension (Df) of TNP aggregates to primary particle size as a function of pH, ionic strength, ion valence, and SRHA. Slope represents sensitivity which was determined from fractal dimension vs. surface areas plot. Details regarding these calculations have been described in Section 3.3.2. | ||
The sensitivity of aggregate morphology to particle surface area, however, showed quite different trend in divalent ions. At pH 7 in CaCl2, an increase in the slope was observed with IS indicating sensitivity of compactness of aggregate to particle size increased with IS in divalent ions. This may be due to higher charge screening from divalent ions leading to more collision opportunities at high IS. Furthermore at pH 10 slope was almost neutral at 10 mM CaCl2, indicating the insensitivity of compactness of aggregate to particle size. However slope decreased from pH 7 to pH 10 at 10 mM CaCl2, a trend opposite to observed in KCl.
The addition of SRHA added complexity to the relationship between aggregate morphology and surface areas. At pH 7 and 10 mM KCl, the slope showed an upward trend in the presence of SRHA indicating that compactness of aggregate was more influenced by surfaces areas in the presence of SRHA. Furthermore the larger slope was observed with additional SRHA, indicating that smaller primary particles showed higher tendency to interact with organic matter, which subsequently influenced aggregate morphology. However with an increase in IS (100 mM KCl) we observed a nearly non-zero slope in the presence of SRHA. The significantly high concentration of KCl diminished the role of primary particle size interactions with SRHA. At pH 7 in CaCl2 slope decreased with addition of SRHA at 10 and 100 mM CaCl2, which was opposite to the trend observed in KCl. However, the addition of SRHA led to a greater slope at pH 10 in CaCl2. At pH 10, KCl with SRHA, slope increased significantly in the presence of SRHA at 10 mM KCl, similar to observed at pH 7 in KCl. Slope increased from pH 7 to pH 10 at 10 mM KCl and CaCl2 in the presence of SRHA, which is also similar trend observed without SRHA.
Overall a linear correlation was observed between fractal dimension and surface areas of primary nanoparticles, which can be useful for the prediction of nanoparticle aggregate fate in the aquatic environment. However, the solution chemistry and presence of organic matter affected the relationship significantly. This may be due to changes in surface charge and aggregate size, which are also function of aquatic chemistry.
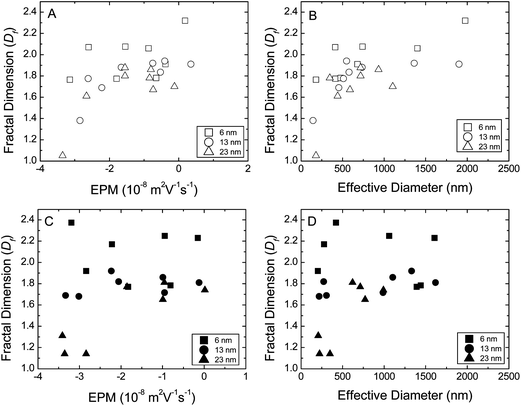 | ||
| Fig. 4 Fractal dimensions (Df) of distinctly sized TNP aggregates as a function of (A) electrophoretic mobilities (EPM) in absence of SRHA, (B) hydrodynamic diameter in absence of SRHA, (C) EPM in presence of SRHA, and (D) hydrodynamic diameter in presence of SRHA. Each data point for fractal dimension, EPM and effective diameter is the average of at least 3 measurements. | ||
Fractal dimension also showed significant dependence on aggregate size (Fig. 4B and D). Df showed a marked increase with aggregate size for all 3 sized TNPs. Fractal dimension of aggregates increased with aggregate size up to a threshold radius where, Df became independent of aggregate size. Beyond this threshold radius, we observed that aggregates comprised of 6 nm had a greater Df than those comprised of 13 nm particles followed by those comprised of 23 nm particle.
Overall both surface charge and aggregate size influenced morphology of TNP aggregates significantly. Influence of surface charge, and aggregate size diminished with reduced primary particle size. This is not surprising given that for small dense NPs electrostatic interactions contribute a smaller percentage to the total interaction energy as particle size decreases. These differences in morphology of TNP aggregates composed of distinctly sized primary particles will have significant effect on the transport of these aggregates in aquatic environment due to change in density, hydrodynamic interactions, surface roughness and strength of aggregates.
4. Summary and conclusions
In this study morphology of nanoparticle aggregates has been correlated with primary particle size and aquatic chemistry. This study shows that there exists a linear relationship between aggregate morphology and surface areas of primary nanoparticles, which can be useful for the prediction of nanoparticle fate in the aquatic environment. However, both solution chemistry, and presence of natural organic matter can notably affect the morphology of nanoparticle aggregate. Our previous study29 showed that primary particle size of nano-TiO2 influenced transport of aggregates through porous media significantly. We found that under straining condition aggregates formed with larger primary nanoparticles (23 nm) can break up more easily than those with smaller ones (6 nm and 13 nm), which subsequently increased the transport of larger nanoparticles. It was revealed from the previous study that the lower fractal dimension of 23 nm-aggregate led to higher break up potential during the transport through porous media. Since the breakage of nanoparticle aggregate is significantly dependent on the aggregate morphology, considerations of these parameters are necessary for future prediction of nanomaterial aggregates in the aquatic environment.Acknowledgements
Funding was provided by the University of California Center for the Environmental Implications of Nanotechnology (National Science Foundation and Environmental Protection Agency under Cooperative Agreement # DBI-0830117). We would like to thank Dr Lutz Mädler, and Dr Suman Pokhrel, University of Bremen, Germany for providing the nanomaterials. Finally, we are grateful for the effort of undergraduate research assistants Jose Valle (Funded by CCRAA #P031C080183-09), and Elizabeth Horstman (funded by National Science Foundation Undergraduate Research and Mentoring grant (MYBEST@UCRDBI-0731660)).Notes and references
- M. Auffan, J. Rose, J.-Y. Bottero, G. V. Lowry, J.-P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634 CrossRef CAS.
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS.
- M. R. Wiesner, G. V. Lowry, P. Alvarez, D. Dionysiou and P. Biswas, Environ. Sci. Technol., 2006, 40, 4336–4345 CrossRef CAS.
- K. L. Chen, S. E. Mylon and M. Elimelech, Langmuir, 2007, 23, 5920–5928 CrossRef CAS.
- K. L. Chen and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7607–7614 CrossRef CAS.
- T. Phenrat, N. Saleh, K. Sirk, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2007, 41, 284–290 CrossRef CAS.
- T. Phenrat, H.-J. Kim, F. Fagerlund, T. Illangasekare and G. V. Lowry, J. Contam. Hydrol., 2010, 118, 152–164 CrossRef CAS.
- D. P. Jaisi, N. B. Saleh, R. E. Blake and M. Elimelech, Environ. Sci. Technol., 2008, 42, 8317–8323 CrossRef CAS.
- C. O. Hendren, X. Mesnard, J. Dröge and M. R. Wiesner, Environ. Sci. Technol., 2011, 45, 2562–2569 CrossRef CAS.
- I. Chowdhury, D. M. Cwiertny and S. L. Walker, Environ. Sci. Technol., 2012, 46, 6868–6976 Search PubMed.
- A. A. Keller, H. Wang, D. Zhou, H. S. Lenihan, G. Cherr, B. J. Cardinale, R. Miller and Z. Ji, Environ. Sci. Technol., 2010, 44, 1962–1967 CrossRef CAS.
- M. A. Kiser, P. Westerhoff, T. Benn, Y. Wang, J. Pérez-Rivera and K. Hristovski, Environ. Sci. Technol., 2009, 43, 6757–6763 CrossRef CAS.
- T. M. Benn and P. Westerhoff, Environ. Sci. Technol., 2008, 42, 4133–4139 CrossRef CAS.
- P. Westerhoff, G. Song, K. Hristovski and M. A. Kiser, J. Environ. Monit., 2011, 13, 1195–1203 RSC.
- K. A. Dunphy Guzman, M. R. Taylor and J. F. Banfield, Environ. Sci. Technol., 2006, 40, 1401–1407 CrossRef CAS.
- C. O. Robichaud, A. E. Uyar, M. R. Darby, L. G. Zucker and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 4227–4233 CrossRef CAS.
- C. O. Robichaud, D. Tanzil, U. Weilenmann and M. R. Wiesner, Environ. Sci. Technol., 2005, 39, 8985 CrossRef CAS.
- K. L. Chen and M. Elimelech, Langmuir, 2006, 22, 10994–11001 CrossRef CAS.
- K. L. Chen, S. E. Mylon and M. Elimelech, Environ. Sci. Technol., 2006, 40, 1516–1523 CrossRef CAS.
- T. Phenrat, H.-J. Kim, F. Fagerlund, T. Illangasekare, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2009, 43, 5079 CrossRef CAS.
- K. A. Dunphy Guzman, M. P. Finnegan and J. F. Banfield, Environ. Sci. Technol., 2006, 40, 7688–7693 CrossRef CAS.
- R. A. French, A. R. Jacobson, B. Kim, S. L. Isley, R. L. Penn and P. C. Baveye, Environ. Sci. Technol., 2009, 43, 1354–1359 CrossRef CAS.
- I. Chowdhury, Y. Hong, R. J. Honda and S. L. Walker, J. Colloid Interface Sci., 2011, 360, 548–555 CrossRef CAS.
- I. Chowdhury, D. M. Cwiertny and S. L. Walker, Environ. Sci. Technol., 2012, 46, 6868–6976 Search PubMed.
- I. Chowdhury and S. L. Walker, J. Colloid Interface Sci., 2012, 369, 16–22 CrossRef CAS.
- B. J. R. Thio, D. Zhou and A. A. Keller, J. Hazard. Mater., 2011, 189, 556–563 CrossRef CAS.
- R. F. Domingos, N. Tufenkji and K. J. Wilkinson, Environ. Sci. Technol., 2009, 43, 1282 CrossRef CAS.
- A. M. Horst, A. C. Neal, R. E. Mielke, P. R. Sislian, W. H. Suh, L. Madler, G. D. Stucky and P. A. Holden, Appl. Environ. Microbiol., 2010, 76, 7292–7298 CrossRef CAS.
- I. Chowdhury, S. Pokhrel, S. Mylon, L. Mädler and S. Walker, unpublished work.
- D. Jassby, J. Farner Budarz and M. Wiesner, Environ. Sci. Technol., 2012, 46, 6934–6941 CrossRef CAS.
- W. Y. Teoh, R. Amal and L. Madler, Nanoscale, 2010, 2, 1324–1347 RSC.
- I. Chowdhury, Y. Hong and S. L. Walker, Colloids Surf., A, 2010, 368, 91–95 CrossRef CAS.
- J. C. Crittenden and H. Montgomery Watson, Water Treatment Principles and Design, J. Wiley, Hoboken, N.J., 2005 Search PubMed.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7963–7969 CrossRef CAS.
- G. C. Bushell, Y. D. Yan, D. Woodfield, J. Raper and R. Amal, Adv. Colloid Interface Sci., 2002, 95, 1–50 CrossRef CAS.
- J. Gregory, Particles in Water: Properties and Processes, IWA Pub.; Taylor & Francis, London; Boca Raton, FL, 2006 Search PubMed.
- J. J. Lenhart, R. Heyler, E. M. Walton and S. E. Mylon, J. Colloid Interface Sci., 2010, 345, 556–560 CrossRef CAS.
- Y. He, J. Wan and T. Tokunaga, J. Nanopart. Res., 2008, 10, 321–332 CrossRef CAS.
- M. Elimelech, J. Gregory, X. Jia and R. A. Williams, Particle Deposition and Aggregation: Measurement, Modeling and Simulation, Butterworth-Heinemann, 1995 Search PubMed.
- K. M. Buettner, C. I. Rinciog and S. E. Mylon, Colloids Surf., A, 2010, 366, 74–79 CrossRef CAS.
- R. Amal, J. Raper and T. D. Waite, J. Colloid Interface Sci., 1992, 151, 244–257 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |