Seasonal and spatial variability in chemical composition and mass closure of ambient ultrafine particles in the megacity of Los Angeles†
Nancy Dahera, Sina Hasheminassaba, Martin M. Shaferb, James J. Schauerb and Constantinos Sioutas*a
aUniversity of Southern California, Department of Civil and Environmental Engineering, Los Angeles, CA, USA. E-mail: sioutas@usc.edu
bUniversity of Wisconsin–Madison, Environmental Chemistry and Technology Program, Madison, WI, USA
First published on 28th November 2012
Abstract
Emerging toxicological research has shown that ultrafine particles (UFP, dp < 0.1–0.2 μm) may be more potent than coarse or fine particulate matter. To better characterize quasi-UFP (PM0.25, dp < 0.25 μm), we conducted a year-long sampling campaign at 10 distinct areas in the megacity of Los Angeles, including source, near-freeway, semi-rural receptor and desert-like locations. Average PM0.25 mass concentration ranged from 5.9 to 16.1 μg m−3 across the basin and over different seasons. Wintertime levels were highest at the source site, while lowest at the desert-like site. Conversely, summertime concentrations peaked at the inland receptor locations. Chemical mass reconstruction revealed that quasi-UFP in the basin consisted of 49–64% organic matter, 3–6.4% elemental carbon, 9–15% secondary ions (SI), 0.7–1.3% trace ions, and 5.7–17% crustal material and trace elements, on a yearly average basis. Organic carbon (OC), a major constituent of PM0.25, exhibited greatest concentrations in fall and winter at all sites, with the exception of the inland areas. Atmospheric stability conditions and particle formation favored by condensation of low-volatility organics contributed to these levels. Inland, OC concentrations peaked in summer due to increased PM0.25 advection from upwind sources coupled with secondary organic aerosol formation. Among SI, nitrate peaked at semi-rural Riverside sites, located downwind of strong ammonia sources. Moreover, ionic balance indicated an overall neutral quasi-UFP aerosol, with somewhat lower degree of neutralization at near-freeway sites in winter. Anthropogenic metals peaked at the urban sites in winter while generally increased at the receptor areas in summer. Lastly, coefficients of divergence analysis showed that while PM0.25 mass is relatively spatially homogeneous in the basin, some of its components, mainly EC, nitrate and several toxic metals, are unevenly distributed. These results suggest that population exposure to quasi-UFP can substantially vary by season and over short spatial scales in the megacity of Los Angeles.
Environmental impactNumerous epidemiological and toxicological studies documented a firm association between increased health risks and exposure to particulate matter (PM). Current regulatory efforts are focused on the reduction of ambient mass levels of PM10 and PM2.5 (dp < 10 and 2.5 μm, respectively). However, increasing epidemiological evidence demonstrated a strong correlation between health endpoints and ultrafine particles (UFP, dp < 0.1–0.2 μm). To improve the characterization of UFP, we conducted a year-long sampling campaign at 10 distinct areas in the megacity of Los Angeles, including source, near-freeway, receptor and desert-dominated locations. The objective was to determine the chemical composition as well as temporal and spatial variability of quasi-UFP (dp < 0.25 μm). Findings of this study provide data to support regulatory agencies as well as epidemiological and toxicity studies. |
1. Introduction
A considerable body of epidemiological and toxicological studies found consistent associations between adverse health effects and exposure to particulate matter (PM), specifically particles less than 10 and 2.5 μm in aerodynamic diameter (PM10 and PM2.5, respectively).1,2 Accordingly, current regulatory efforts are focused on the reduction of ambient mass levels of PM10 and PM2.5. However, mounting epidemiological evidence3–5 demonstrated a strong correlation between health endpoints and ultrafine particles (UFP, typically defined as particles with dp < 0.1–0.2 μm6).In urban areas, UFP are primarily emitted from motor vehicles but can also be formed through gas-to-particle atmospheric conversion processes.7–9 Despite their very low contribution to overall PM mass, ultrafine particles dominate ambient particle number concentrations.10 UFP also have a large surface area relative to fine or coarse particles11 and a high pulmonary deposition efficiency.12 Because of their increased number and surface area, ultrafine particles can carry substantial amounts of toxic air pollutants, such as organic carbon (OC), polycyclic aromatic hydrocarbons (PAHs) and transition metals, many of which have been implicated in inducing inflammatory effects.13,14 Some of these PM components, such as PAHs and OC, dominate the ultrafine fraction.15,16 Thus, UFP may have significant health responses; greater than- or independent of the effects induced by larger size particles, as shown by previous studies which reported that UFP exhibited a greater mass-based redox activity than fine and coarse particles.15,17,18 The redox activity strongly correlated with the OC and PAHs content of PM, in particular, which was highest in the UFP samples.
Contrarily to coarse (2.5 < dp < 10 μm) and fine (PM2.5) PM, which have distinct sources, a major fraction of accumulation particles (>UFP in diameter and <2.5 μm) originates from UFP. Consequently, there is no clear cut-point that separates ultrafine from accumulation mode PM. While UFP are conventionally defined as PM accounting for greater than 80% of total particle number concentrations,10 aerosol number median diameters can be as large as 90–150 nm in some polluted urban areas in summer.7,8 Therefore, the majority of number-based ambient particles may not always lie below a cut-point of 100 nm. Furthermore, ultrafine soot particles have agglomerate-like structures,19 which are characterized by their large surface area and low density compared to equivalent spherical particles. A considerable fraction of these fractal particles would be classified by an inertial separator as ultrafine because of their low density, whereas a particle mobility-based classifier would classify them in the accumulation mode range due to their large surface area.6,20,21 Thus, the cut-point separating accumulation and ultrafine PM can be ambiguous, varying with location and season.6 The current study focuses on particles with aerodynamic diameter less than 0.25 μm. We will be referring to these particles as quasi-UFP hereafter.
Lastly, unlike PM2.5 or PM10, for which there is an abundance of continuous and time-integrated mass or chemically speciated data, significantly less is known about the spatial and seasonal variation of ultrafine particles as well as their chemical composition. Efforts in this direction include investigations of the characteristics of ambient UFP in European cities.22–24 However, little equivalent data is available for areas in the U.S. Determining these PM properties is important in assessing human exposure to ultrafine particles. A recent cohort study conducted in the LAB reported positive associations of blood biomarkers of inflammation and ambulatory ST-segment depression with quasi-UFP, but no associations with larger accumulation-mode PM0.25–2.525 and PM2.5–10.25,26 Such differences in systemic responses between particle-size fractions, notably accumulation and ultrafine mode of regulated fine PM, highlight the importance of characterizing particles in the size range <0.25 μm, particularly in impacted communities of the LAB. Previous studies conducted in this area investigated PM2.5 and PM10, with a few exploring UFP properties.11,27–29 Moreover, measurements were mostly restricted to few sampling sites and/or limited sampling intervals (few days or weeks). These studies therefore do not provide insight on the larger scale distribution of quasi-UFP in the complex Los Angeles (LA) air shed. To expand the existing knowledge on quasi-ultrafine particles, we conducted a year-long sampling campaign at 10 distinctly different areas in the LAB, including source, near-freeway, semi-rural receptor and desert-like locations. The objective of this study is to provide the much-needed information on the chemical composition as well as seasonal and spatial variability of quasi-UFP. Findings of this work could provide the basis for designing epidemiological and toxicity studies to mitigate population exposure to quasi-ultrafine particles.
2. Methodology
2.1. Sampling sites
To characterize quasi-UFP, 10 sampling sites, impacted by different PM sources and representing distinct areas of the Los Angeles Basin, were selected and set up. These included source, near-freeway, semi-rural receptor and desert-like areas. A three letter code is used to designate the sites throughout this manuscript. Their location, along with nearby major freeways, is shown in Fig. 1. Detailed description of the sampling locations and selection criteria is provided in a separate publication.30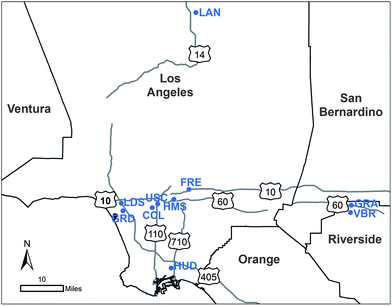 | ||
| Fig. 1 Location of the sampling sites. | ||
Briefly, the 10 sites can be classified according to their geographical location into Long Beach (HUD), western LA (GRD, LDS), central LA (CCL, USC), eastern LA (HMS, FRE), Riverside County (VBR, GRA) and Lancaster (LAN); in order of their increasing distance from the coast. The Long Beach site, HUD, is located about 2 km inland of the ports of LA and Long Beach in a mixed residential and industrial area. It is also immediately to the east of Terminal Island Freeway (ca. 100 m) and 1.2 km west of I-710, both of which have a high fleet and fraction of heavy duty diesel vehicles (HDDV) (up to 25% on I-710 (ref. 31)) serving to transfer cargo from the ports to inland locations.30,31 Because of its surrounding environment and location, HUD is considered as a source site in the LAB. In contrast, Riverside sites (VBR and GRA), which are located in a mostly rural-agricultural area about 80 km inland from downtown LA, represent pollutant receptor areas in the basin. Given their location on the prevalent air trajectory crossing the LAB from coast to inland, these sites are considerably affected by advected, aged and photochemically processed PM from upwind regions.8,30 Furthermore, while both sites are relatively close to major streets, GRA is in the vicinity of CA-60 freeway (∼300 m north). GRA may therefore be more significantly impacted by freeway emissions than VBR. Further inland across the basin, LAN represents a typical desert-like site, located in a rural region of Lancaster city to the north of the LAB. This monitoring site is over 2 km away from the nearest freeway (CA-14), but near local arterial roadways.30,32 The 6 remaining sites, situated in the LA area, represent urban sites. The western LA sites (GRD and LDS) correspond to coastal areas, with LDS immediately to the southwest of I-405 freeway. The eastern LA (HMS, FRE) and central USC sites are located within 50–800 m of nearby freeways. Moreover, although not located next to a freeway, CCL is adjacent to surface streets with significant motor vehicle traffic.
2.2. Sample collection
Twenty-four-hour time-integrated samples were concurrently collected at the 10 sampling sites for a year-long period extending from April 2008 to March 2009. Samples were collected on a weekly basis during weekdays. Two parallel Sioutas Personal Cascade Impactor Samplers33,34 (Sioutas™ PCIS, SKC Inc., Eighty Four, PA, USA), operating at 9 lpm, were deployed at each site to collect size-fractionated particles in the following size ranges: <0.25 μm (quasi-UFP), 0.25–2.5 μm, and 2.5–10 μm. However, the present study only focuses on the quasi-UFP fraction. The coarse PM chemical composition is discussed in detail in other publications.35–38 For the purpose of chemical analyses, one PCIS was loaded with Teflon filters (Pall Life Sciences, Ann Arbor, MI) while the other one with quartz microfiber filters (Whatman International Ltd, Maidstone, England). 37 mm filters were used as collection media for the quasi-ultrafine particles.2.3. Sample analysis
Mass concentration of the quasi-UFP samples was determined by pre- and post-weighing the 24 hour time-integrated Teflon filters using a Microbalance (Mettler Toledo Inc., Columbus, OH, USA), following equilibrium under controlled temperature and relative humidity conditions (22–24 °C and 40–50%, respectively).To perform chemical analyses of the PM0.25 samples, the collected Teflon and quartz filters were sectioned into portions. Elemental and organic carbon (EC and OC, respectively) measurements were performed on individual quartz filter portions while all other analyses were conducted on monthly composites of sectioned filters. Water-soluble inorganic ions and total elements were quantified from monthly composites. EC and OC contents of the filters were determined using the NIOSH Thermal Optical Transmission method.39 The ionic content of the filters was quantified by ion chromatography (IC)40 following water extraction and filtration of the samples. The elemental mass of the filter substrates was measured using a high resolution magnetic sector Inductively Coupled Plasma Mass Spectrometry (Thermo-Finnigan Element 2).41 A mixed-acid (nitric acid, hydrochloric acid and hydrofluoric acid) microwave-aided digestion was applied for extraction of total elements. Lastly, concentration values that were below the limit of detection (<2 × total analytical uncertainty) were assumed as half the detection limit.
3. Meteorology
Select meteorological parameters, organized by sites' clusters and season, are listed in Table S1.† Parameters include mean temperature, relative humidity, precipitation as well as vector-average wind speed and direction. Data was acquired from the online database of the California Air Resources Board. Mean temperature and relative humidity exhibited more seasonality in the inland areas at Riverside and Lancaster, compared to Long Beach and urban LA. Across the air basin, temperatures were coolest in winter while warmest in summer. Mean temperatures were also relatively warm in fall, spanning a range of 18.8–22 °C across the sites' clusters. These meteorological conditions are typical of the LAB, where the warm period extends to September and October.8 Mean relative humidity was highest in the coastal areas (Long Beach and west LA) and reached peak values in summer (76.8 and 81%). Furthermore, excluding Lancaster, humidity levels increased in summer in the basin. Lancaster, on the other hand, was characterized by an intense seasonal variability in its temperature and relative humidity. Its average temperature varied from a low of 8.7 °C in winter to a high of 27.9 °C in summer. Its relative humidity also dropped from a maximum of 55.9% in winter to a minimum of 26.9% in summer and was generally substantially lower than levels at the other sites. These extreme meteorological patterns are consistent with the desert-like and distinct nature of LAN. At all sites, total precipitation was negligible in summer and spring but greatest in fall and winter. Additionally, wind speeds were generally stronger away from the coast, with more calms (<0.5 m s−1) observed in fall and winter. Wind speed during spring and summer was also higher than during fall and winter. Moreover, wind direction was consistent with the prevailing air trajectory crossing the LAB from coast to inland.42 Wind blowing from the coast was predominantly westerly or southwesterly during summer and spring, resulting in strong onshore flows that transport pollutants across the basin. In winter, the wind shifted and had a predominantly northerly component across the sites' clusters, with the exception of Lancaster. In Lancaster, winds mainly originated from the west irrespective of the season.4. Results and discussion
In the following sections, all reported seasonal concentrations correspond to arithmetic averages with seasons segregated into spring (March–May), summer (June–August), fall (September–November) and winter (December–February). Respective errors represent one standard error, unless otherwise noted. Moreover, all 10 sampling sites presented in figures and tables of this manuscript are arranged from coastal to inland (HUD, GRD, LDS, CCL, USC, HMS, FRE, VBR, GRA and LAN).4.1. Particulate mass
Seasonal-average mass concentration of quasi-UFP in the Los Angeles Basin is presented in Table 1 at each of the sampling sites. As can be inferred, the mass levels of quasi-UFP spanned a broad range of 5.9–16.1 μg m−3 across the seasons and basin. Spatially, the mass concentration in winter, when emissions from local primary sources predominate, was greatest at HUD. This site is located in a pollutant source region of the LAB close to I-710 and Terminal Island freeways, as well as industrial sources and ports of Los Angeles and Long Beach.30 On the other hand, the wintertime concentration was lowest at the desert-like LAN site due to its remote location from urban sources. In contrast, concentrations were highest at receptor GRA and LAN sites in warm summer or also spring seasons, likely due to increased secondary aerosol formation and particle advection from upwind “source” areas. The strong and predominantly westerly/southwesterly wind in spring and summer (Table S1†) transports primary PM and gaseous precursors from the western to the eastern edge of the basin,43,44 resulting in an elevation in concentration and a photochemically processed aerosol at the inland locations. Similar observations have been extensively reported for the receptor area of Riverside.7,8,29 Among urban LA sites, concentrations were lowest at the western/coastal sites (GRD, LDS).| Long Beach | Western LA | Central LA | Eastern LA | Riverside | Lancaster | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| HUD | GRD | LDS | CCL | USC | HMS | FRE | VBR | GRA | LAN | |
| Spring | 7.9 ± 0.8 | 6 ± 0.7 | 5.4 ± 0.7 | 6.6 ± 0.5 | 9.2 ± 1.4 | 7.4 ± 0.7 | 7.7 ± 0.8 | 8.8 ± 1.2 | 10.0 ± 1.7 | 7.5 ± 1.3 |
| Summer | 9.4 ± 0.5 | 7.1 ± 0.5 | 7.0 ± 0.4 | 7.6 ± 0.6 | 8.7 ± 1.4 | 9.4 ± 1.1 | 10.4 ± 0.8 | 9.7 ± 0.7 | 12.6 ± 0.9 | 13.2 ± 1.9 |
| Fall | 13 ± 1.6 | 10.7 ± 1.2 | 9.9 ± 1.0 | 11.2 ± 1.1 | 13.3 ± 1.4 | 11.4 ± 1.2 | 12.6 ± 1.4 | 14.9 ± 2.0 | 13.1 ± 1.4 | 8.8 ± 0.9 |
| Winter | 16.1 ± 2.8 | 8.4 ± 1.0 | 8.5 ± 0.9 | 10 ± 0.9 | 11.4 ± 1.4 | 9.1 ± 0.8 | 9.7 ± 0.9 | 9.8 ± 1.4 | 10.3 ± 1.2 | 5.9 ± 0.7 |
Seasonally, a pronounced variability was observed at HUD and LAN. At source site HUD, peak concentrations occurred in winter and fall (16.1 ± 2.8 and 13 ± 1.6 μg m−3, respectively) as opposed to spring and summer (7.9 ± 0.8 and 9.4 ± 0.5 μg m−3, respectively). Higher atmospheric stability conditions, enhanced particle formation by condensable vapors, freshly emitted from vehicles8 and peak cargo activity in the fall30 likely contributed to the elevated levels. At LAN, the mass concentration varied from a low of 5.9 ± 0.7 μg m−3 in winter to a high of 13.2 ± 1.9 μg m−3 in summer. As aforementioned, this trend can be associated with increased eastward PM advection and secondary aerosol formation in summer which offset atmospheric dilution effects. Conversely, peak levels were generally measured in fall at urban LA and Riverside sites. This pattern, which is in agreement with findings of Sardar et al.,29 could be attributed to limited particle dispersion due to the low wind speeds (≤1.6 m s−1, Table S1†) combined with extended secondary PM formation into early fall and particle formation from directly emitted condensable vapors.8 Secondary aerosol production is also supported by the mean peak levels of ozone, an indicator of photochemical activity, which had appreciable concentrations in early fall (Fig. S1†). Moreover, the low temperature coupled with increased relative humidity and stagnant conditions in the evening and early night in fall may enhance the partitioning of low vapor pressure compounds to the particle phase compared to spring–summer, as observed in earlier investigations conducted in this basin.31,45 These combined effects lead to more pronounced concentrations in fall relative to the other seasons.
4.2. Mass reconstruction
To determine the bulk composition of quasi-ultrafine particles in the LAB, a chemical mass reconstruction was conducted at each of the sampling sites during spring, summer, fall and winter, as shown in Fig. 2a–d. PM chemical species were grouped into organic matter (OM), EC, secondary ions (SI), trace ions as well as metals, including crustal material (CM) and trace elements (TE). To account for the contributions of non-carbon atoms (e.g. oxygen, hydrogen) to the total mass of OM, a factor of 1.6 ± 0.2 and 2.1 ± 0.2 is recommended for urban and non-urban aerosols, respectively.46 In this study, a factor of 1.8 is used to convert OC to OM for all sites and seasons. SI included ammonium, nitrate and sulfate, while trace ions comprised chloride, sodium and potassium. The proportion of CM in quasi-UFP was determined by summing the oxides of crustal metals, Al, K, Fe, Ca, Mg, Ti and Si, using the following equation:47–49| CM = 1.89Al + 1.21K + 1.43Fe + 1.4Ca + 1.66Mg + 1.67Ti + 2.14Si | (1) |
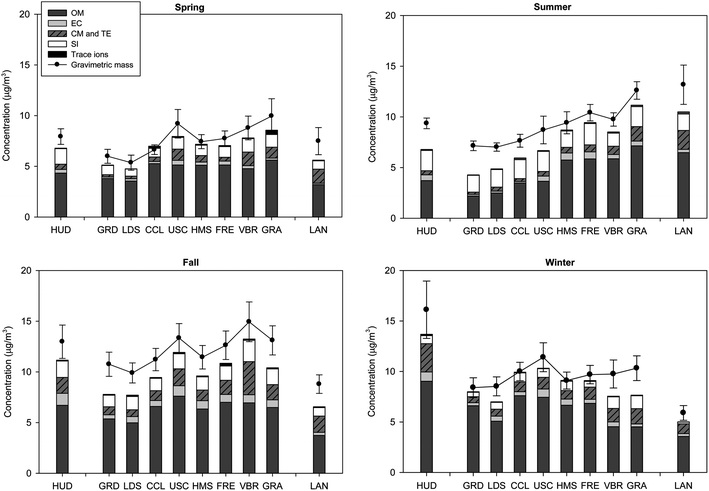 | ||
| Fig. 2 (a–d) Chemical composition and gravimetric mass concentration of quasi-UFP (dp < 0.25 μm) by site in (a) spring; (b) summer, (c) fall and (d) winter. Error bars correspond to one standard error. | ||
Si concentration was estimated as 3.41 × Al (ref. 48) since it was not measured in this study. Lastly, TE consisted of the remaining measured elements such as Cu, Zn and Cr.
On a yearly average basis, the chemical composition of quasi-UFP in the LAB consisted of 49–64% OM, 3–6.4% EC, 9–14.5% SI (5–8.6% sulfate, 0.5–3.8% nitrate and 2.6–5.4% ammonium), 0.9–1.3% trace ions and 5.7–17% CM and TE, as shown in Table 2. These proportions are within the ranges previously reported for ultrafine particles in Southern California.28,29,50 As can be deduced, OM is the dominant constituent of PM0.25, accounting for close to half of its mass. SI and metals were next most abundant, contributing to quasi-UFP in similar amounts.
| OM | EC | SO2−4 | NO−3 | NH+4 | Trace ions | CM + TE | |
|---|---|---|---|---|---|---|---|
| Long beach | 52.6 ± 5 | 6.4 ± 0.8 | 8.6 ± 1.3 | 0.5 ± 0.1 | 5.4 ± 1 | 0.8 ± 0.1 | 10 ± 1.7 |
| Western LA | 54.6 ± 4 | 3.8 ± 0.4 | 8.1 ± 0.9 | 0.8 ± 0.2 | 5.2 ± 0.6 | 0.7 ± 0.1 | 5.7 ± 0.6 |
| Central LA | 58.9 ± 2.8 | 5 ± 0.5 | 7.8 ± 1 | 1.7 ± 0.3 | 4.7 ± 0.6 | 1.3 ± 0.3 | 8.6 ± 0.9 |
| Eastern LA | 63.8 ± 2.2 | 5.9 ± 0.4 | 7.2 ± 0.7 | 0.9 ± 0.1 | 4.8 ± 0.5 | 1.2 ± 0.2 | 8.2 ± 0.9 |
| Riverside | 51.4 ± 1.5 | 4 ± 0.3 | 5.4 ± 0.6 | 3.8 ± 0.6 | 4 ± 0.3 | 1.3 ± 0.3 | 13.4 ± 1 |
| Lancaster | 48.9 ± 3.1 | 3.1 ± 0.4 | 5 ± 0.9 | 1.3 ± 0.2 | 2.6 ± 0.5 | 0.9 ± 0.1 | 16.8 ± 1.6 |
The agreement between the gravimetric and reconstructed mass concentrations is overall good as indicated by the ratio of reconstructed to gravimetric mass, which respectively averaged 0.87 ± 0.02, 0.80 ± 0.03, 0.83 ± 0.02 and 0.88 ± 0.02 during spring, summer, fall and winter across all sites. These ratios are in the range of 23–40% and 6–45% of unidentified material, reported in other studies for PM0.1 and quasi-UFP in the LAB.11,51 This agreement between the two PM mass concentrations is further corroborated by applying a least squares linear regression to the average gravimetric and reconstructed mass concentrations of monthly samples from all sites, as displayed in Fig. S2a–d.† As can be seen, the coefficient of determination (R2) and slope respectively spanned a range of 0.71–0.82 and 0.77–1.01 across the seasons, indicating a good agreement between the two mass concentrations. This agreement, however, exhibited a seasonal dependence, with a higher fraction of undetermined particulate mass observed during the warm summer season in consistence with other studies.29,48,52 This fraction, which was highest at the coastal (27–40%) and inland LAN (20%) sites, could be primarily associated with the uncertainty in the OC multiplication factor. As aforementioned, a factor of 1.8 is used for all sites and seasons in this study. Nevertheless, this factor may vary with location, season and time of day.46 Applying the same multiplication factor for all sites and seasons may introduce some uncertainty in the estimation of the organic mass. OM may be underestimated in summer, when the aerosol is composed of photochemically processed and advected particles. Secondary organic aerosol (SOA) constituents are more oxygenated and polar than primary aerosol components, resulting in an increase in the average organic molecular weight to carbon weight ratio (MWt/C Wt).46 Furthermore, as the aerosol ages during air mass transit, its composition may be altered due to the addition of semi-volatile products of photochemical reactions, leading also to an increase in MWt/C Wt.46 In addition, the fraction of unaccounted mass could also be related to uncertainties in the multiplication factors used to convert metals to oxides and un-estimated aerosol-bound water, especially for the coastal sites.
4.3. Chemical composition
Elemental carbon. EC, a key tracer of diesel exhaust,53 was a minor constituent of PM0.25, accounting for 3–6.4% of its mass with levels ranging from 0.13 to 1.17 μg m−3 across the seasons and basin, as displayed in Table 2 and Fig. 3a–d. EC levels and contribution to quasi-UFP (∼3%) were generally lowest at remote LAN site, which is 2 km away from the nearest freeway. Low levels were also measured at community-based (away from freeway) GRD and CCL sites as well as coastal LDS, particularly in spring–summer when values were mostly close or below the detection limit. In contrast, the largest EC contribution (6.4 ± 0.8%) to PM0.25 occurred at source site HUD due to its close proximity to freeways with high volume of HDDV in port service.30,31 Accordingly, HUD exhibited highest EC concentrations across the basin during fall (1.17 ± 0.16 μg m−3) and winter (0.92 ± 0.18 μg m−3), when emissions from local primary sources dominate. Fall- and winter-time EC levels were next most dominant at USC in central LA (1.02 ± 0.16 and 0.82 ± 0.12 μg m−3, respectively) followed by HMS in eastern LA (0.81 ± 0.12 and 0.60 ± 0.13 μg m−3, respectively). These sites are located in close-to-modest proximity (within 150–800 m) to heavily trafficked freeways. In urban LA and Long Beach, EC concentrations were highest during stagnant fall or also winter seasons compared to warmer summer and spring. Similarly, EC concentration peaked in fall at Riverside. However, notable is that comparable/greater EC levels were measured in summer compared to winter at receptor VBR/GRA sites. Given the greater atmospheric dilution of primary emissions in summer, these higher summertime concentrations of primary species such as EC are mainly attributed to the increased advection of air parcels from upwind sources in the western part of the basin.
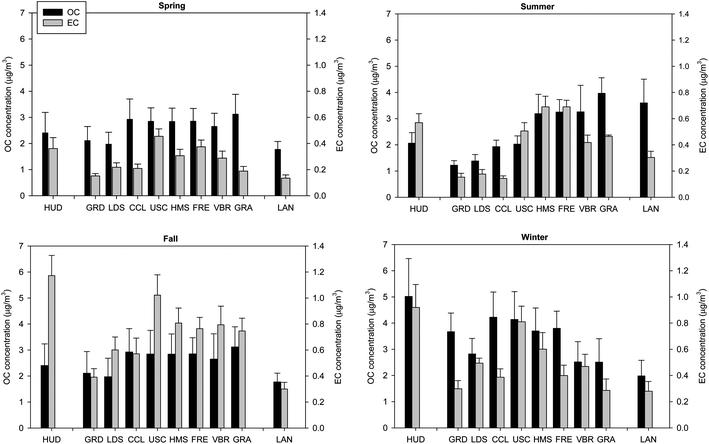 | ||
| Fig. 3 (a–d) Average concentration of organic carbon (OC) and elemental carbon (EC) in quasi-UFP (dp < 0.25 μm) by site in (a) spring, (b) summer, (c) fall and (d) winter. Error bars correspond to one standard error. | ||
Lastly, it is noteworthy that EC concentrations significantly declined compared to earlier studies in this basin. Current PM0.25 EC levels at Riverside are about 15 and 54% lower than those reported by Fine et al.27 in PM0.18 during summer and winter, respectively. Additionally, quasi-ultrafine EC levels at USC and Long Beach are 30 and 48% lower than those reported by Minguillón et al.54 during March–May, respectively. These reductions in EC concentration reflect improved diesel engine emission controls and the effectiveness of the Clean Trucks Program of Long Beach and Los Angeles ports (San Pedro Bay Ports Clean Air Action Plan).
Organic matter. OM was the predominant component in quasi-UFP throughout the seasons, accounting for 49–64% of its mass on a yearly average basis, as presented in Table 2. Remote LAN site exhibited the lowest fraction while central (CCL, USC) and eastern LA sites (FRE, HMS) displayed the greatest proportions. The high fractions possibly reflect contributions from vehicular emissions, which constitute a major source of ultrafine particles in urban areas of LA.29
OM can be directly emitted from primary sources, such as fossil fuel combustion, or produced in the atmosphere via SOA formation processes.55 To evaluate the extent of primary and secondary contributions to OM, the seasonal variation of OC and OC/EC ratio were investigated as illustrated in Fig. 3a–d and Table 3 at each of the sampling sites. EC, which predominantly originates from combustion processes, is used as a tracer of primary combustion-generated OC.55 OC concentrations were higher in fall and winter compared to spring and summer at all locations, with the exception of Riverside (VBR, GRA) and LAN where noticeable peaks were apparent in summer. With the exception of GRD and CCL, where EC levels were mostly close to the detection limit, the elevated winter- and fall-time concentrations were overall accompanied by low OC/EC ratios compared to spring or summer. These ratios, however, exceeded those commonly reported for urban areas,56,57 likely due to the very low EC levels relative to OC, particularly in winter. Nonetheless, the seasonality in OC/EC ratio and stronger OC–EC correlation in fall–winter (Table 3) indicate that OC is of predominately primary origin during fall and winter. The elevated fall- and winter-time OC concentrations can be attributed to atmospheric stability conditions and favored particle formation or growth by condensable organics freshly emitted from vehicles8 as well as SOA formation driven by the low mixing height,58 to a much lesser extent. Wintertime OC concentration varied from ∼2 to 5 μg m−3 across the basin, with greater levels measured in the urban areas where local primary sources are more abundant than inland. In particular, wintertime OC concentrations were greatest at source site HUD and central USC, highlighting the importance of primary combustion emissions at these near-freeway sites. Conversely, OC levels were highest at receptor Riverside (VBR, GRA) and LAN sites in summer, averaging 3.3–4 μg m−3. These levels, which followed a similar pattern to mass and exceeded wintertime concentrations, can be attributed to a combination of increased PM advection and SOA formation in summer, as documented in previous studies.8,43 SOA production at these locations, enhanced by the accumulation of organic gases in the advected air parcel during transit from coast to inland,43 is supported by the significant summertime increase in OC/EC ratio, which varied from 5.9–9.4 in fall–winter to 8.4–14.6 in summer at these locations, as well as low OC–EC correlations (R = 0.36–0.41). Moreover, excluding CCL and GRD where EC levels were close to the detection limit in spring–summer, OC/EC ratios were greatest at the semi-rural/rural areas (VBR, GRA, LAN) compared to the urban sites, reflecting the importance of SOA formation at these inland locations.
| Long Beach | Western LA | Central LA | Eastern LA | Riverside | Lancaster | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HUD | GRD | LDS | CCL | USC | HMS | FRE | VBR | GRA | LAN | ||
| OC/EC | Spring | 9.9 ± 2.6 | 14.7 ± 2.2 | 10.1 ± 1 | 16.3 ± 2.2 | 8.8 ± 2.4 | 13.7 ± 2.8 | 11.7 ± 3.1 | 13.5 ± 2.8 | 19.9 ± 3.2 | 14 ± 1.5 |
| Summer | 4.1 ± 0.5 | 9.1 ± 0.9 | 9.3 ± 1 | 15.5 ± 1.3 | 4.9 ± 1 | 6.6 ± 1.9 | 5.1 ± 0.8 | 8.4 ± 0.9 | 10 ± 1.9 | 14.6 ± 1.9 | |
| Fall | 3.9 ± 0.8 | 7.9 ± 0.9 | 5.6 ± 0.7 | 7.9 ± 1.6 | 4.4 ± 0.4 | 5.3 ± 0.8 | 4.9 ± 0.4 | 5.9 ± 0.8 | 5.1 ± 0.4 | 9.4 ± 1.5 | |
| Winter | 6.9 ± 1.3 | 19.6 ± 4.6 | 5.9 ± 0.7 | 15.9 ± 4.3 | 5.3 ± 0.4 | 5.7 ± 0.8 | 5.9 ± 1 | 5.8 ± 0.8 | 8.2 ± 1.6 | 9.8 ± 2.3 | |
| R(OC–EC) | Spring–summer | 0.44 | — | — | — | 0.34 | 0.37 | 0.43 | 0.36 | 0.41 | — |
| Fall–winter | 0.54 | 0.25 | 0.71 | 0.72 | 0.78 | 0.77 | 0.53 | 0.80 | 0.56 | 0.44 | |
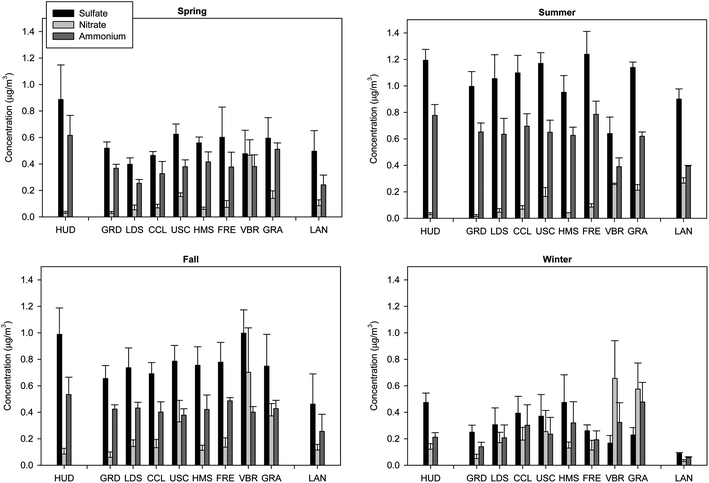 | ||
| Fig. 4 (a–d) Average concentration of sulfate, nitrate and ammonium in quasi-UFP (dp < 0.25 μm) by site in (a) spring, (b) summer, (c) fall and (d) winter. Error bars correspond to one standard error. | ||
In addition, an ion balance was conducted by determining concentration ratios (in charge equivalents) of the amount of measured ammonium to the amount of ammonium required to fully neutralize measured sulfate and nitrate, as plotted in Fig. 5a–d for all sampling sites on a seasonal-average basis. A value of 1 suggests that sulfate and nitrate are fully neutralized by ammonium in the form of ammonium nitrate and ammonium sulfate. A value below 1 implies that particles are partially acidic, mostly in the form of ammonium bisulfate.66 As can be inferred, ammonium is generally present in sufficient amounts to fully neutralize sulfate and nitrate, with some values exceeding 1. The latter indicate the presence of excess ammonium, possibly bound to organic acids.67 With the exception of GRA and LAN, lower ionic ratios were observed in fall and winter than spring and summer at the sampling sites. This trend implies a lesser amount of excess ammonium in fall and winter, consistent with an increased formation of ammonium nitrate during cooler periods. Furthermore, ratios in fall and summer were overall lower at the semi-rural/rural inland locations (VBR, GRA, LAN) than urban areas, with ratios dropping below unity at VBR (0.79 ± 0.21) and LAN (0.93 ± 0.16) in fall and summer, respectively. These results indicate a somewhat lower degree of neutralization at these inland receptor sites and a slightly acidic aerosol at VBR and LAN, possibly due to long-range transport of gaseous acidic precursors. In contrast, ratios were highest at semi-rural GRA in winter, when PM advection is limited, which can be attributed to the greater ammonia emissions in this area, resulting in increased aerosol neutralization. On the other hand, wintertime ratios were lowest and approaching unity at source site HUD in Long Beach and urban central USC in winter, implying a lesser degree of neutralization likely due to their close proximity to heavily trafficked freeways. It is possible that ammonia cannot fully access sulfuric acid because of organic coatings on the sulfate core or sulfate could be interacting with EC, as argued in previous investigations at USC site.50 Thus, ammonia preferentially reacts with larger size accumulation mode particles.50
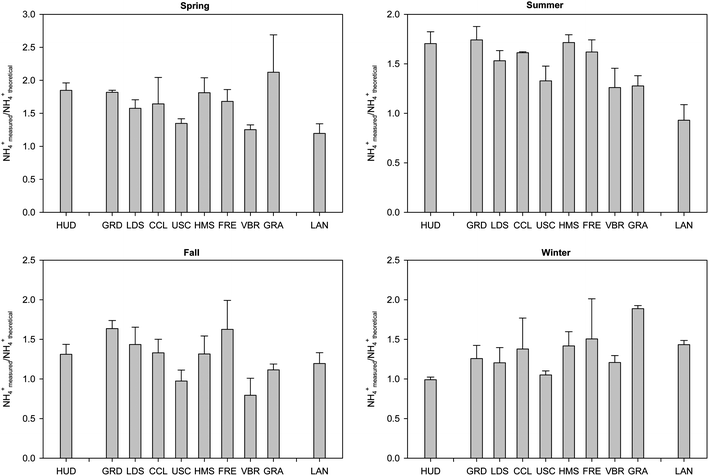 | ||
| Fig. 5 (a–d) Average molar concentration ratio of measured ammonium to theoretical ammonium required for complete neutralization of nitrate and sulfate ions in (a) spring, (b) summer, (c) fall and (d) winter. Error bars represent one standard error. | ||
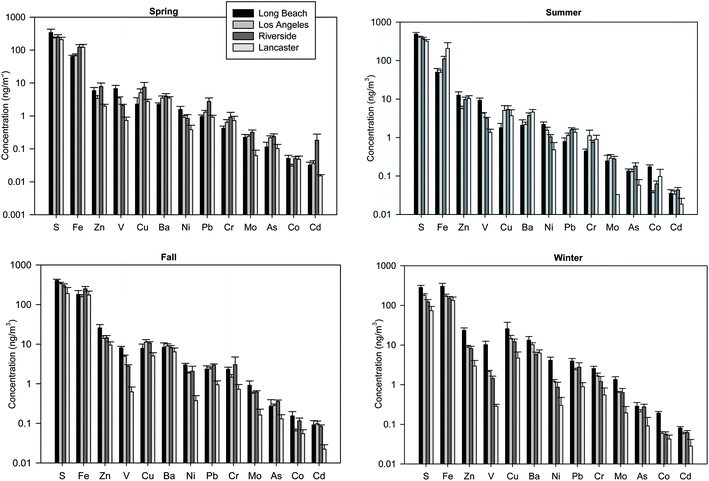 | ||
| Fig. 6 (a–d) Average concentration of metals in quasi-UFP (dp < 0.25 μm) by site in (a) spring, (b) summer, (c) fall and (d) winter. Error bars represent one standard error. | ||
4.4. Spatial variability
To determine the spatial variation of PM0.25 in the Los Angeles Basin, coefficients of divergence (COD) were calculated between sites' pairs for quasi-UFP mass and its main components. COD provides information on the degree of uniformity between sites, necessary to determine the density of monitoring networks required to accurately assess personal exposure to PM in large metropolitan areas. The COD for a given site pair is defined as follows: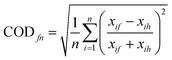 | (2) |
The CODs were estimated between site pairs using monthly concentrations, as illustrated in Fig. 7. Overall, findings showed a fairly homogeneous spatial variability for quasi-UFP mass but heterogeneous distribution for some of its components, mainly EC, CM, select toxic metals and nitrate. These results imply that differences in source contributions may account for the divergence observed for some of PM0.25 species. The low median COD value (0.15) and narrow interquartile range (of the order of 0.1 units) for mass indicate its relatively spatial homogeneity throughout the year. The prevalence of similar local sources at most of the sampling locations, namely vehicular emissions, as well as regional PM transport across the basin contribute to this limited variability among sites. The maximum observed COD of 0.30 is associated with sites' pairs including the distinct desert-like LAN site. Accordingly, the median COD for OC, largest contributor to quasi-UFP, equaled 0.20, indicating its moderately homogeneous spatial distribution. OC is attributed to primary combustion sources and SOA formation processes. The latter are regional in nature, resulting in a homogenous dispersion of OC.80 On the other hand, high heterogeneity is observed for EC, which displayed a median COD of 0.35. This disparity among sites can be related to their varying proximity to freeways and dissimilar HDDV traffic density. Similarly, CM and specific toxic metals were unevenly distributed, in agreement with their unequal abundance at the urban and inland sites, as discussed earlier. A great heterogeneity (median COD > 0.3) is observed for elements of industrial emissions (Cr) and vehicular emissions (Fe, Zn, Cu, Ba, Mo, Co, As, Cd and Pb) because their concentration heavily depends on their proximity to sources and traffic patterns. V, an element of bunker-fuel combustion from marine vessels, also exhibited an uneven distribution in accordance with its decreasing concentration from coast to inland. Similarly, a strong heterogeneity was noted for Ni, which originated from oil combustion in Long Beach whereas industrial sources at the remaining sites, as argued earlier. S, on the other hand, was relatively homogenous across the basin (median COD = 0.18), consistent with its partly secondary origin. Moreover, a very strong variability is observed for nitrate throughout the year (median COD = 0.61), which can be largely associated with differences in the source strength of its precursor gases at the sites, particularly Riverside. Contrarily, sulfate and ammonium displayed a modest heterogeneity (median COD = 0.22, 0.23) but high maxima (>0.45). The latter were driven by the low wintertime levels at remote LAN, due to its distinct meteorology and lack of acidic gaseous precursors.
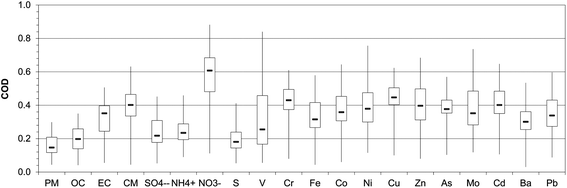 | ||
| Fig. 7 Coefficient of divergence (COD) between sites pairs across the year. Box plots show the minimum, first quartile, median, third quartile and maximum observed values. | ||
These results suggest that the variability in some of quasi-UFP chemical constituents on “inter-community” scales in the LAB may lead to considerably variable population exposure to quasi-ultrafine particles in this basin. It is, however, important to acknowledge that these results are based on 24 hour time-integrated data and do not reflect the spatial distribution in quasi-UFP over finer time scales. Previous studies conducted in eastern and source regions of the LAB examined the spatial variability in particle number concentrations on diurnal scales, using hourly CODs.45,81 Their findings showed greater spatial heterogeneity during morning commute and late evening hours. Such variability, which is relevant to acute exposures, may not be fully captured by this time-integrated approach.
5. Summary and conclusions
To provide insight on the spatial variability in chemical composition of quasi-UFP, a year-long sampling campaign was conducted at 10 distinct areas in the Los Angeles Basin. Chemical mass closure showed that the composition of PM0.25 is in the range of 49–64% OM, 3–6.4% EC, 9–15% SI, 0.7–1.3% trace ions, and 5.7–17% CM and TE, on a yearly average basis. Quasi-UFP, which mostly consisted of OC, varied from 5.9 to 16.1 μg m−3 across the sites and seasons. In winter, mass and OC levels were higher in the urban areas, specifically at source site HUD, where local primary sources are more abundant than inland. These elevated concentrations can be attributed to atmospheric stability conditions and favored particle formation by condensable organic vapors. Conversely, summertime levels peaked at inland Riverside and LAN sites due to increased SOA formation and PM0.25 advection from upwind polluted urban areas. EC levels were highest at HUD due to its proximity to freeways with a large fleet of HDDV while minimal at the community-based and desert-like sites. Among SI, sulfate and ammonium displayed summertime maxima due to increased photochemical activity. In contrast, nitrate levels dropped from a high in winter–fall to a low in summer–spring, reflecting the partitioning of ammonium nitrate to its gaseous precursors with increasing temperature. Furthermore, an ion balance revealed a generally neutral aerosol, with lesser degree of neutralization at near-freeway HUD and USC sites. Furthermore, toxic metals of anthropogenic origin overall peaked at urban LA and Long Beach sites in stagnant winter while receptor Riverside and Lancaster in summer. Lastly, inter-urban variability was investigated by calculating COD between sites' pairs. Results showed that while quasi-UFP mass is relatively spatially homogeneous in the basin, some of its chemical components, mainly EC, nitrate as well as CM and several toxic metals, were unevenly distributed.In conclusion, findings of this study indicate that the composition and concentration of quasi-UFP are considerably affected by location and season. The spatial and temporal variability of quasi-ultrafine particles should thus be considered in the design of epidemiological studies in order to accurately assess population exposure to PM in complex urban air sheds, such as the megacity of Los Angeles.
Acknowledgements
This study was funded by the South Coast Air Quality Management District (SCAQMD) (award #11527). We wish to acknowledge the support of USC Provost's Ph.D. fellowship. We also would like to thank the staff at the Wisconsin State Laboratory of Hygiene for their assistance with the chemical analyses.References
- R. McConnell, K. Berhane, F. Gilliland, S. J. London, H. Vora, E. Avol, W. J. Gauderman, H. G. Margolis, F. Lurmann, D. C. Thomas and J. M. Peters, Environ. Health Perspect., 1999, 107, 757–760 CrossRef CAS.
- C. A. Pope III, JAMA, J. Am. Med. Assoc., 2002, 287, 1132–1141 CrossRef.
- R. J. Delfino, C. Sioutas and S. Malik, Environ. Health Perspect., 2005, 113, 934–946 CrossRef.
- A. Peters, H. E. Wichmann, T. Tuch, J. Heinrich and J. Heyder, Am. J. Respir. Crit. Care Med., 1997, 155, 1376–1383 CAS.
- P. Penttinen, K. L. Timonen, P. Tiittanen, A. Mirme, J. Ruuskanen and J. Pekkanen, Environ. Health Perspect., 2001, 109, 319–323 CrossRef CAS.
- C. Sioutas, R. J. Delfino and M. Singh, Environ. Health Perspect., 2005, 113, 947–955 CrossRef.
- S. B. Sardar, P. M. Fine, P. R. Mayo and C. Sioutas, Environ. Sci. Technol., 2004, 39, 932–944 CrossRef.
- S. Kim, S. Shen and C. Sioutas, J. Air Waste Manage. Assoc., 1995, 2002(52), 297–307 Search PubMed.
- D. Westerdahl, S. Fruin, T. Sax, P. M. Fine and C. Sioutas, Atmos. Environ., 2005, 39, 3597–3610 CrossRef CAS.
- L. Morawska, S. Thomas, N. Bofinger, D. Wainwright and D. Neale, Atmos. Environ., 1998, 32, 2467–2478 CrossRef CAS.
- L. S. Hughes and G. R. Cass, Environ. Sci. Technol., 1998, 32, 1153–1161 CrossRef CAS.
- D. C. Chalupa, P. E. Morrow, G. n. Oberdörster, M. J. Utell and M. W. Frampton, Environ. Health Perspect., 2004, 112, 879–882 CrossRef CAS.
- Y. Kumagai, T. Arimoto, M. Shinyashiki, N. Shimojo, Y. Nakai, T. Yoshikawa and M. Sagai, Free Radical Biol. Med., 1997, 22, 479–487 CrossRef CAS.
- P. H. N. Saldiva, R. W. Clarke, B. A. Coull, R. C. Stearns, J. Lawrence, G. G. K. Murthy, E. Diaz, P. Koutrakis, H. Suh, A. Tsuda and J. J. Godleski, Am. J. Respir. Crit. Care Med., 2002, 165, 1610–1617 CrossRef.
- N. Li, C. Sioutas, A. Cho, D. Schmitz, C. Misra, J. Sempf, M. Wang, T. Oberley, J. Froines and A. Nel, Environ. Health Perspect., 2003, 111, 455–460 CrossRef CAS.
- A. Eiguren-Fernandez, A. H. Miguel, P. A. Jaques and C. Sioutas, Aerosol Sci. Technol., 2003, 37, 201–209 CrossRef CAS.
- A. K. Cho, C. Sioutas, A. H. Miguel, Y. Kumagai, D. A. Schmitz, M. Singh, A. Eiguren-Fernandez and J. R. Froines, Environ. Res., 2005, 99, 40–47 CrossRef CAS.
- L. Ntziachristos, J. Froines, A. Cho and C. Sioutas, Part. Fibre Toxicol., 2007, 4, 5 CrossRef.
- S. Friedlander, Dynamics of Agglomerate Formation and Restructuring in Smoke, Dust and Haze, Oxford University Press, New York, 2000 Search PubMed.
- P. H. McMurry, X. Wang, K. Park and K. Ehara, Aerosol Sci. Technol., 2002, 36, 227–238 CrossRef CAS.
- M. Geller, S. Biswas and C. Sioutas, Aerosol Sci. Technol., 2006, 40, 709–723 CrossRef CAS.
- A. G. Allen, E. Nemitz, J. P. Shi, R. M. Harrison and J. C. Greenwood, Atmos. Environ., 2001, 35, 4581–4591 CrossRef CAS.
- J. Ruuskanen, T. Tuch, H. Ten Brink, A. Peters, A. Khlystov, A. Mirme, G. P. A. Kos, B. Brunekreef, H. E. Wichmann, G. Buzorius, M. Vallius, W. G. Kreyling and J. Pekkanen, Atmos. Environ., 2001, 35, 3729–3738 CrossRef CAS.
- T. A. Pakkanen, V.-M. Kerminen, C. H. Korhonen, R. E. Hillamo, P. i. Aarnio, T. Koskentalo and W. Maenhaut, Atmos. Environ., 2001, 35, 4593–4607 CrossRef CAS.
- R. J. Delfino, N. Staimer, T. Tjoa, D. L. Gillen, A. Polidori, M. Arhami, M. T. Kleinman, N. D. Vaziri, J. Longhurst and C. Sioutas, Environ. Health Perspect., 2009, 117, 1232–1238 CrossRef CAS.
- R. J. Delfino, D. L. Gillen, T. Tjoa, N. Staimer, A. Polidori, M. Arhami, C. Sioutas and J. Longhurst, Environ. Health Perspect., 2011, 119, 196–202 CrossRef CAS.
- P. M. Fine, B. Chakrabarti, M. Krudysz, J. J. Schauer and C. Sioutas, Environ. Sci. Technol., 2004, 38, 1296–1304 CrossRef CAS.
- G. R. Cass, L. A. Hughes, P. Bhave, M. J. Kleeman, J. O. Allen, L. G. Salmon, G. R. Cass, L. A. Hughes, P. Bhave, M. J. Kleeman, J. O. Allen and L. G. Salmon, Philos. Trans. R. Soc., A, 2000, 358, 2581–2592 CrossRef CAS.
- S. B. Sardar, P. M. Fine, P. R. Mayo and C. Sioutas, Environ. Sci. Technol., 2005, 39, 932–944 CrossRef CAS.
- P. Pakbin, N. Hudda, K. L. Cheung, K. F. Moore and C. Sioutas, Aerosol Sci. Technol., 2010, 44, 514–525 CrossRef CAS.
- K. Moore, M. Krudysz, P. Pakbin, N. Hudda and C. Sioutas, Aerosol Sci. Technol., 2009, 43, 587–603 CrossRef CAS.
- K. F. Moore, V. Verma, M. C. Minguillón and C. Sioutas, Aerosol Sci. Technol., 2010, 44, 526–540 CrossRef CAS.
- C. Misra, M. Singh, S. Shen, C. Sioutas and P. M. Hall, J. Aerosol Sci., 2002, 33, 1027–1047 CrossRef CAS.
- M. Singh, P. A. Jaques and C. Sioutas, Atmos. Environ., 2002, 36, 1675–1689 CrossRef CAS.
- P. Pakbin, N. Hudda, K. L. Cheung, K. F. Moore and C. Sioutas, Aerosol Sci. Technol., 2010, 44, 514–525 CrossRef CAS.
- P. Pakbin, Z. Ning, M. M. Shafer, J. J. Schauer and C. Sioutas, Aerosol Sci. Technol., 2011, 45, 949–963 CrossRef CAS.
- K. Cheung, M. R. Olson, B. Shelton, J. J. Schauer and C. Sioutas, Atmos. Environ., 2012, 59, 1–10 CrossRef CAS.
- K. Cheung, N. Daher, W. Kam, M. M. Shafer, Z. Ning, J. J. Schauer and C. Sioutas, Atmos. Environ., 2011, 45, 2651–2662 CrossRef CAS.
- M. E. Birch and R. A. Cary, Analyst, 1996, 121, 1183–1190 RSC.
- E. A. Stone, C. J. Hedman, R. J. Sheesley, M. M. Shafer and J. J. Schauer, Atmos. Environ., 2009, 43, 4205–4213 CrossRef CAS.
- Y. Zhang, J. J. Schauer, M. M. Shafer, M. P. Hannigan and S. J. Dutton, Environ. Sci. Technol., 2008, 42, 7502–7509 CrossRef CAS.
- L. S. Hughes, J. O. Allen, P. Bhave, M. J. Kleeman, G. R. Cass, D. Y. Liu, D. P. Fergenson, B. D. Morrical and K. A. Prather, Environ. Sci. Technol., 2000, 34, 3058–3068 CrossRef CAS.
- S. N. Pandis, R. A. Harley, G. R. Cass and J. H. Seinfeld, Atmos. Environ., Part A, 1992, 26, 2269–2282 CrossRef.
- J. O. Allen, L. S. Hughes, L. G. Salmon, P. R. Mayo, R. J. Johnson and G. R. Cass, Characterization and Evolution of Primary and Secondary Aerosols During PM2. 5 and PM10 Episodes in the South Coast Air Basin, 2000 Search PubMed.
- N. Hudda, K. Cheung, K. F. Moore and C. Sioutas, Atmos. Chem. Phys., 2010, 10, 11385–11399 CrossRef CAS.
- B. J. Turpin and H.-J. Lim, Aerosol Sci. Technol., 2001, 35, 602–610 CAS.
- J. C. Chow, J. G. Watson, E. M. Fujita, Z. Lu, D. R. Lawson and L. L. Ashbaugh, Atmos. Environ., 1994, 28, 2061–2080 CrossRef CAS.
- C. Hueglin, R. Gehrig, U. Baltensperger, M. Gysel, C. Monn and H. Vonmont, Atmos. Environ., 2005, 39, 637–651 CrossRef CAS.
- G. M. Marcazzan, S. Vaccaro, G. Valli and R. Vecchi, Atmos. Environ., 2001, 35, 4639–4650 CrossRef CAS.
- Z. Ning, M. D. Geller, K. F. Moore, R. Sheesley, J. J. Schauer and C. Sioutas, Environ. Sci. Technol., 2007, 41, 6000–6006 CrossRef CAS.
- M. Arhami, M. Sillanpää, S. Hu, M. R. Olson, J. J. Schauer and C. Sioutas, Aerosol Sci. Technol., 2009, 43, 145–160 CrossRef CAS.
- K. F. Ho, S. C. Lee, J. J. Cao, J. C. Chow, J. G. Watson and C. K. Chan, Sci. Total Environ., 2006, 355, 276–287 CrossRef CAS.
- J. J. Schauer, J. Exposure Sci. Environ. Epidemiol., 2003, 13, 443–453 CrossRef CAS.
- M. C. Minguillón, M. Arhami, J. J. Schauer and C. Sioutas, Atmos. Environ., 2008, 42, 7317–7328 CrossRef.
- B. J. Turpin, J. J. Huntzicker, S. M. Larson and G. R. Cass, Environ. Sci. Technol., 1991, 25, 1788–1793 CrossRef CAS.
- L. M. Castro, C. A. Pio, R. M. Harrison and D. J. T. Smith, Atmos. Environ., 1999, 33, 2771–2781 CrossRef CAS.
- B. J. Turpin and J. J. Huntzicker, Atmos. Environ., 1995, 29, 3527–3544 CrossRef CAS.
- R. Strader, F. Lurmann and S. N. Pandis, Atmos. Environ., 1999, 33, 4849–4863 CrossRef CAS.
- L. S. Hughes, J. O. Allen, L. G. Salmon, P. R. Mayo, R. J. Johnson and G. R. Cass, Environ. Sci. Technol., 2002, 36, 3928–3935 CrossRef CAS.
- M. J. Mysliwiec and M. J. Kleeman, Environ. Sci. Technol., 2002, 36, 5376–5384 CrossRef CAS.
- M. Mozurkewich, Atmos. Environ., Part A, 1993, 27, 261–270 CrossRef.
- S. B. Sardar, P. M. Fine and C. Sioutas, J. Geophys. Res., [Atmos.], 2005, 110, 14 CrossRef.
- L. S. Hughes, J. O. Allen, M. J. Kleeman, R. J. Johnson, G. R. Cass, D. S. Gross, E. E. Gard, M. E. Gälli, B. D. Morrical, D. P. Fergenson, T. Dienes, C. A. Noble, D.-Y. Liu, P. J. Silva and K. A. Prather, Environ. Sci. Technol., 1999, 33, 3506–3515 CrossRef CAS.
- M. D. Geller, P. M. Fine and C. Sioutas, J. Air Waste Manage. Assoc., 1995, 2004(54), 1029–1039 Search PubMed.
- M. D. Geller, S. Kim, C. Misra, C. Sioutas, B. A. Olson and V. A. Marple, Aerosol Sci. Technol., 2002, 36, 748–762 CrossRef CAS.
- Q. Zhang, C. O. Stanier, M. R. Canagaratna, J. T. Jayne, D. R. Worsnop, S. N. Pandis and J. L. Jimenez, Environ. Sci. Technol., 2004, 38, 4797–4809 CrossRef CAS.
- E. Dinar, T. Anttila and Y. Rudich, Environ. Sci. Technol., 2008, 42, 793–799 CrossRef CAS.
- S. Becker, L. A. Dailey, J. M. Soukup, S. C. Grambow, R. B. Devlin and Y.-C. T. Huang, Environ. Health Perspect., 2005, 113, 1032–1038 CrossRef CAS.
- J. Isakson, T. A. Persson and E. Selin Lindgren, Atmos. Environ., 2001, 35, 3659–3666 CrossRef CAS.
- G. Lu, J. R. Brook, M. Rami Alfarra, K. Anlauf, W. Richard Leaitch, S. Sharma, D. Wang, D. R. Worsnop and L. Phinney, Atmos. Environ., 2006, 40, 2767–2782 CrossRef CAS.
- P. G. Sanders, N. Xu, T. M. Dalka and M. M. Maricq, Environ. Sci. Technol., 2003, 37, 4060–4069 CrossRef CAS.
- B. D. Garg, S. H. Cadle, P. A. Mulawa, P. J. Groblicki, C. Laroo and G. A. Parr, Environ. Sci. Technol., 2000, 34, 4463–4469 CrossRef CAS.
- L. Ntziachristos, Z. Ning, M. D. Geller, R. J. Sheesley, J. J. Schauer and C. Sioutas, Atmos. Environ., 2007, 41, 5684–5696 CrossRef CAS.
- C.-C. Lin, S.-J. Chen, K.-L. Huang, W.-I. Hwang, G.-P. Chang-Chien and W.-Y. Lin, Environ. Sci. Technol., 2005, 39, 8113–8122 CrossRef CAS.
- A. Thorpe and R. M. Harrison, Sci. Total Environ., 2008, 400, 270–282 CrossRef CAS.
- G. C. Lough, J. J. Schauer, J.-S. Park, M. M. Shafer, J. T. DeMinter and J. P. Weinstein, Environ. Sci. Technol., 2004, 39, 826–836 CrossRef.
- Z. Xie, L. Sun, J. D. Blum, Y. Huang and W. He, J. Geophys. Res., [Atmos.], 2006, 111, 11 Search PubMed.
- J. Sternbeck, Å. Sjödin and K. Andréasson, Atmos. Environ., 2002, 36, 4735–4744 CrossRef CAS.
- J. G. Wilson, S. Kingham, J. Pearce and A. P. Sturman, Atmos. Environ., 2005, 39, 6444–6462 CrossRef CAS.
- J. R. Turner and D. T. Allen, J. Air Waste Manage. Assoc., 1995, 2008(58), 196–215 Search PubMed.
- M. Krudysz, K. Moore, M. Geller, C. Sioutas and J. Froines, Atmos. Chem. Phys., 2009, 9, 1061–1075 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2em30615h |
| This journal is © The Royal Society of Chemistry 2013 |