
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperparamagnetic iron oxide nanoparticles functionalized by peptide nucleic acids†
Marco Gallia,
Andrea Guerrinib,
Silvia Cauteruccioa,
Pramod Thakarea,
Davide Dovaa,
Francesco Orsinic,
Paolo Arosioc,
Claudio Carraraa,
Claudio Sangregoriode,
Alessandro Lascialfarice,
Daniela Maggioni *ae and
Emanuela Licandro*ae
*ae and
Emanuela Licandro*ae
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy. E-mail: daniela.maggioni@unimi.it
bLA.M.M. c/o Dipartimento di Chimica, Università degli Studi di Firenze, Via della Lastruccia 3, 50019 Sesto F.no (FI), Italy
cDipartimento di Fisica, Università degli Studi di Milano, Via Celoria 20, 20133 Milano, Italy
dICCOM – C.N.R., via Madonna del Piano 10, 50019 Sesto F.no (FI), Italy
eConsorzio INSTM, via G. Giusti 9, 50121, Firenze, Italy
First published on 9th March 2017
Abstract
A novel efficient method has been developed for covalently linking Peptide Nucleic Acid (PNA) oligomers and superparamagnetic iron oxide nanoparticles (SPION), to produce water soluble hybrid nanomaterials that can act as MRI contrast agents, as hyperthermia promoters and as PNA carriers. The multistep procedure involves: (i) preparation of oleate-stabilized SPION by using the thermal decomposition method, to control the size of the magnetic core (here 17 ± 2 nm, by TEM measurement); (ii) exchange of the oleate layer by dimercaptosuccinic acid (DMSA), to impart water solubility and to provide functional groups for PNA grafting; (iii) functionalization of a PNA oligomer with a terminal maleimide moiety, to allow SPION–PNA conjugation by thiol-maleimide Michael addition reaction, exploiting the SH groups of DMSA on the SPION surface. The method was tested using a model PNA decamer containing all four nucleobases (–CTAGATCACT–). SPION–PNA conjugation by SH addition was found more efficient than conjugation through amide bond between the COOH groups of DMSA and the terminal NH2 groups of PNA. Elemental analysis, UV-Vis and IR spectra, and ζ-potential measures confirmed the PNA binding (a loading of ca. 400 PNA strands per SPION was estimated, molar ratio ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 with respect to DMSA). A detailed characterization of the morphology, relaxivity and magnetic properties of the SPION used for PNA binding is reported, and compared to the one relative to the SPION–PNA conjugate. The analysis of the magnetic behaviour showed that the nanoparticles are in the superparamagnetic regime at room temperature, and have a considerably high saturation magnetization (85 emu g−1). Magnetic hyperthermia measurements gave a remarkable value of the heat released (SAR = 65 W g−1), which makes these SPION suitable for magnetic hyperthermia treatments. The SPION were also able to effectively shorten both longitudinal and transverse relaxation times of water, with r1 and r2 values higher with respect to routinely used commercial contrast agents, at the typical fields of clinical instrumentation.
:15 with respect to DMSA). A detailed characterization of the morphology, relaxivity and magnetic properties of the SPION used for PNA binding is reported, and compared to the one relative to the SPION–PNA conjugate. The analysis of the magnetic behaviour showed that the nanoparticles are in the superparamagnetic regime at room temperature, and have a considerably high saturation magnetization (85 emu g−1). Magnetic hyperthermia measurements gave a remarkable value of the heat released (SAR = 65 W g−1), which makes these SPION suitable for magnetic hyperthermia treatments. The SPION were also able to effectively shorten both longitudinal and transverse relaxation times of water, with r1 and r2 values higher with respect to routinely used commercial contrast agents, at the typical fields of clinical instrumentation.
1. Introduction
The high current interest in the uses of multifunctional superparamagnetic iron oxide nanoparticles (SPION) in nanomedicine1 derives from their sensing, moving and heating abilities, imparted by their unique magnetic properties. The most common application is the use as contrast agents for magnetic resonance imaging (MRI).1b,2 In addition, in the presence of SPION, by means of an external ac magnetic field it is also possible to induce local release of heat to the surrounding tissues, which can be exploited as a cancer therapy treatment called Magnetic Fluid Hyperthermia (MFH).3 Currently, MFH is under clinical testing associated with radiotherapy for the treatment of several tumours.4 Other important applications consist in cell labelling, separation and tracking5 and delivery of drugs and genes.6 The last application is particularly useful for preserving labile therapeutic species or for improving their bioavailability.Peptide nucleic acids (PNAs)7 are synthetic polyamide mimics of natural DNA and RNA, in which the (desoxy)ribose-phosphate backbone is replaced by N-(2-aminoethyl)glycyl neutral repeating units (Chart 1). The nucleobases, linked to the backbone through methylencarbonyl linkers, lie at the right distance to pair with the nucleobases of natural DNA or RNA single strand, with high affinity and sequence specificity.
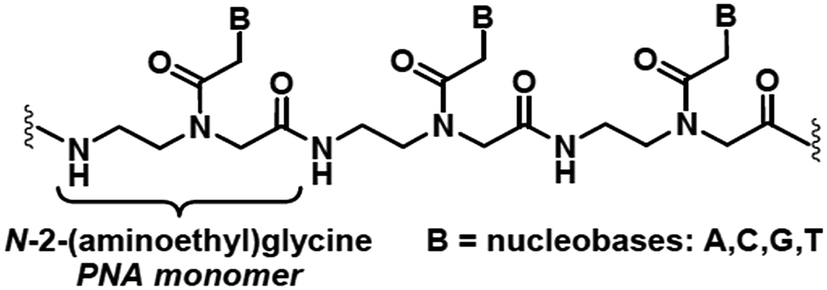 | ||
| Chart 1 A peptide nucleic acid repeating fragment. | ||
The major advantages of PNAs are the resistance to enzymatic biodegradation by proteases and nucleases, and the higher chemical/thermal stability over their duplexes with DNA or RNA strands with respect to dsDNA and dsRNA. These features make PNAs an attractive tool for gene therapy, where the poor in vivo stability of nucleic acids and their ineffective uptake by target cells hamper translation to the clinic of effective platforms developed in in vitro cell assays. To pursue this end, however, it is necessary to enhance PNA bioavailability by conjugation to additional agents. Indeed, PNA major drawbacks are poor solubility in physiological media and very low ability to pass the cellular membranes.
The conjugation of PNA strands to superparamagnetic iron oxide nanoparticles could address these drawbacks, improving for example PNA solubility and cell permeability and, in a more general sense, improving its potential in biology and medicine. In fact, the system SPION–PNA can be suitable for gene analysis in diagnostics and gene therapy by taking advantage, on one hand, of ability of PNA to target in a very specific manner DNA and RNA sequences, and on the other hand by exploiting the superparamagnetic properties of nanoparticles, whose potential in biomedicine is well established. Very few studies appeared in the literature on the binding of PNA to magnetic iron oxide nanoparticles.8,9 In the most recent report 9 the covalent binding was obtained by a multistep procedure, involving functionalization of the surface of naked magnetite nanoparticles with 3-aminopropyltriethoxysilane, followed by reaction with acryloyl chloride, to give acrylamide-grafted SPION, radical polymerization and further covalent functionalization of the polymer coating with streptavidin. Pyrrolidinyl PNA previously functionalized with biotin was finally conjugated to the nanoparticles, via specific biotin–streptavidin interaction. A versatile and effective synthetic platform for grafting PNA oligomers to naked commercial maghemite NPs had been previously developed,8 based on the high affinity of siloxane group for iron oxide. However, the solubility of the PNA-based hybrid nanostructures obtained by this method was not satisfactory.8 Here we describe a novel synthetic strategy, which gives access to hybrid inorganic–organic systems made of iron oxide nanoparticles and PNA strands, for applications as contrast agents for MRI and heat mediators in MFH. The method here reported combines easiness and efficacy with the possibility to tune the SPION size, and allows the obtainment of magnetic PNA with good water solubility.
2. Results and discussion
2.1 Synthesis of the iron oxide nanoparticles
The synthesis of highly monodisperse magnetic iron oxide nanoparticles was extensively addressed in the last years by many research teams. The discovery of thermal decomposition methods enabled a dramatic increase of monodispersion,10 but reproducibility issues are not yet entirely overcome. Actually, many factors affect the size and the crystallinity, and then the magnetic properties of the sample, such as the precursor nature, the heating ramp, the aging time and temperature, the solvent, the molar ratio between the iron precursor and the surfactant.Here we adopted a synthetic methodology in which the iron(0) precursor Fe(CO)5 is decomposed in high-boiling 1-octadecene, in the presence of oleic acid (OA) as capping agent.11 The literature procedure used a 4:1 OA:Fe molar ratio that gave spherical SPION with an average diameter of 10 nm. It is known that the SPION size can be tuned by varying the relative concentrations of iron precursor and capping agent.12,13 In line with this, on increasing the OA:Fe ratio (see Table 1) we found a concomitant increase both of the hydrodynamic diameter (measured by dynamic light scattering analyses, DLS, Fig. S1 ESI†) and of the diameter measured by transmission electron microscopy (TEM, Fig. S2 in ESI†). The size increment was dramatically sensitive to OA:Fe ratio, as can be clearly observed in the graphs reported in Fig. S3 (ESI†). As expected, the mean particle size distributions measured by DLS were significantly larger than the ones obtained by TEM measurements, since DLS returns a hydrodynamic diameter that takes into account not only the magnetic core, but also the OA shell, and the corona shell of solvent interacting with OA, that jointly fluctuates by Brownian motions.
:Fe molar ratios, as measured by DLS (hydrodynamic diameters, intensities distribution) and by TEM
| Sample | OA:Fe molar ratio |
DLS (intensities) (nm) | TEM (nm) |
|---|---|---|---|
| a The population with a larger size (78 nm) observable in Fig. S1 (ESI) represented only a very minor fraction of the whole sample (1.5%, according to the DLS number distribution).b A population with a smaller size (13.5 ± 3.4 nm) was also observed by TEM, probably due to uncompleted Ostwald ripening (Fig. S2, ESI). | |||
| SPION6 | 6:1 |
23.3 ± 1.6a | 11.9 ± 3.0 |
| SPION7 | 7:1 |
26.6 ± 3.0 | 17.0 ± 1.9 |
| SPION8 | 8:1 |
38.95 ± 9.2 | 29.9 ± 2.6b |
The SPION7 sample, obtained with an OA:Fe ratio equal to 7, consisted in highly spherical monodisperse nanoparticles with a mean diameter of 17.0 nm. This sample was chosen for carrying on the work, because its size grants for the highest hyperthermic efficiency among the series of samples. Moreover, the other two samples exhibited several shortcomings (polydispersion, polycrystalline/amorphous structure), which are detrimental for their magnetic properties. Furthermore, the size of SPION7 allows reaching stability once dispersed in water (see below), avoiding possible aggregation favoured by the high magnetic moment of bigger nanoparticles, the last effect highly undesirable for biomedical applications. Therefore, the core of all the nanosystems described hereafter was constituted by the SPION7 sample, even if not explicitly stated.
In order to impart the necessary water solubility and biocompatibility, the oleate layer was exchanged with dimercaptosuccinic acid (DMSA). Actually nanoparticles coated by DMSA are stabilized over a wide pH range by the electrostatic repulsion arising from the negative charge present on their surface even at relatively low pH values (pKa of DMSA: 2.71, 3.43, 9.65, 12.05).14 Besides providing SPION stability in physiological conditions, DMSA offers the further benefit of two functional groups (COOH and SH), which both can be exploited for the covalent bonding of a variety of organic molecules.15
To perform the ligand exchange we used a literature methodology16 (see Scheme 1), modified by the use of sonication during the reaction, which allowed a significant shortening of the exchange time (40 min vs. 12–24 h of the literature procedures), while maintaining the quasi-quantitative recovery of the water-dispersed SPION.
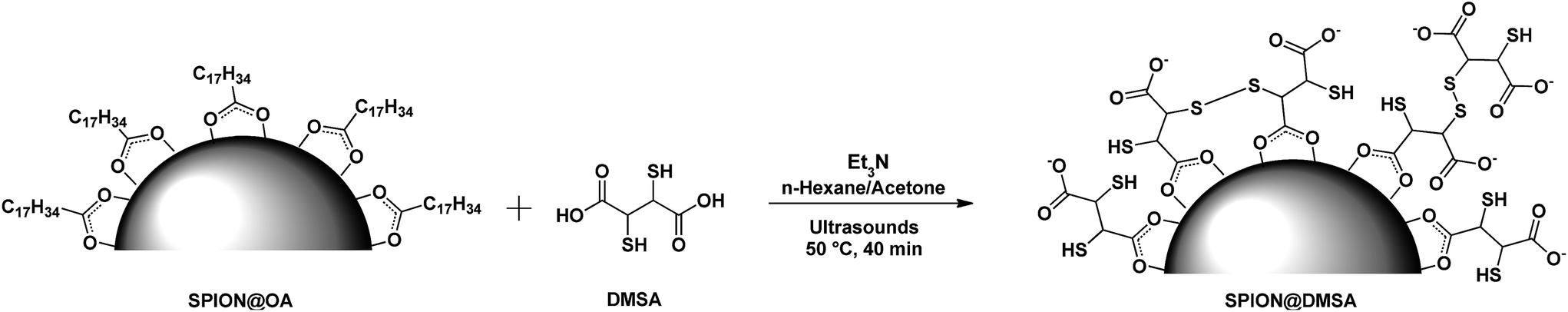 | ||
| Scheme 1 The ligand exchange procedure affording the SPION@DMSA here used for PNA conjugation. Different binding modes of the carboxylate groups on the magnetite surface are depicted in the scheme,16 taking into account also the possibility of oxidative coupling of SH groups.17 | ||
The so obtained SPION@DMSA suspensions, stocked at 4 °C under N2 at pH > 9, were very stable for several months. The ζ-potential (Fig. S4, ESI†) was negative even at quite low pH, as expected from the above reported pKa values. DLS measurements performed on samples at pH 9 showed (Fig. S5, ESI†) in the distribution size graph (intensities) two distinct peaks centred at 146 ± 42 nm and 44 ± 8 nm, indicating a certain degree of aggregation. However, the hydrodynamic distribution by numbers showed that the aggregated population represented only a minority of the whole sample of suspended particles (2.5%).
The morphology of DMSA-stabilized SPION was investigated by Tapping Mode Atomic Force Microscopy (TM-AFM), which showed that the nanoparticles maintained the spherical-like shape observed before ligand exchange (see Fig. 1a). By analysis of several images, the overall size (magnetic core and DMSA shell coating) was estimated as 20.6 ± 2.0 nm (averaged value over 40 observations), in the same range as observed by TEM for precursor SPION@OA, and significantly smaller than the value measured by DLS for SPION@DMSA, due to the contribution to the hydrodynamic diameter of the solvating water that surrounds the NPs.
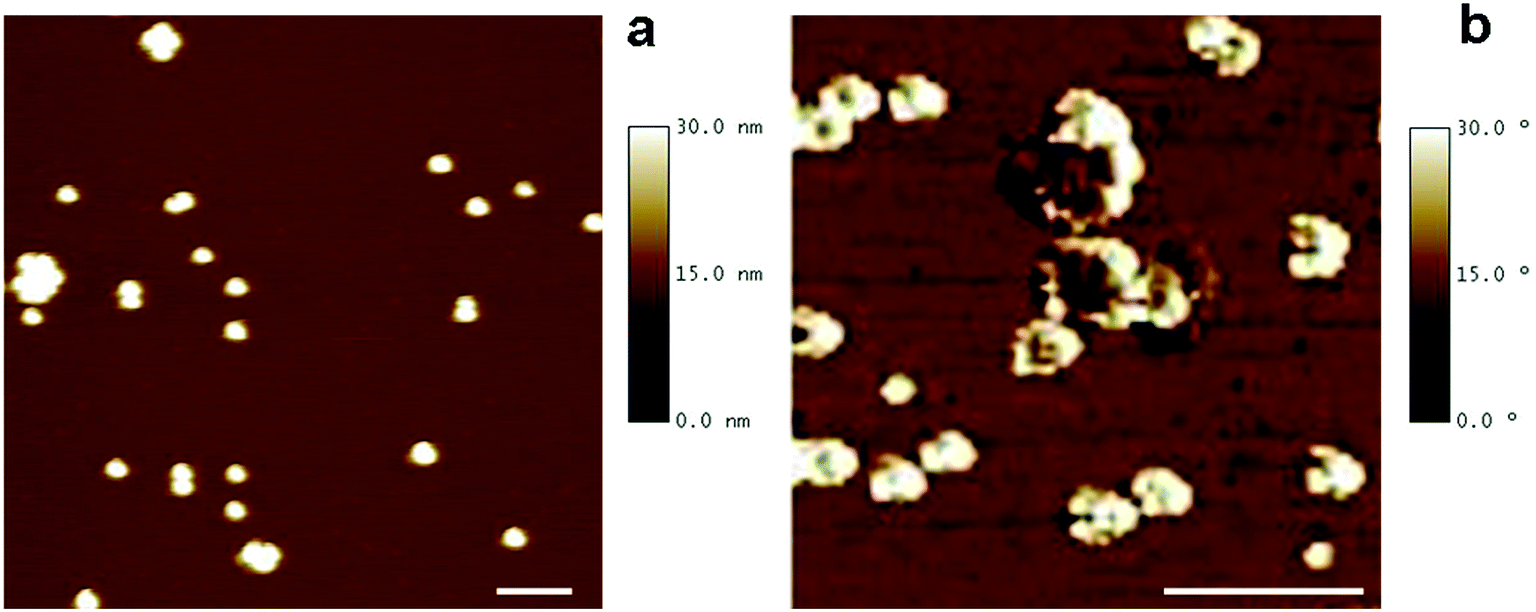 | ||
| Fig. 1 AFM images of SPION@DMSA samples adsorbed on a mica support. (a) AFM topography image. Besides single SPION, some small aggregates are also observed, most likely formed in the deposition step. Scan area 800 × 800 nm2. Vertical scale 30 nm. (b) AFM phase image, which allows the visualization of the coating (white rings) and of the NP magnetic core (darker areas, partially eclipsed by the external coating). Scan area 300 × 300 nm2. For both the images, scale bar = 100 nm. | ||
AFM phase images have also been acquired, to evidence the coating shell surrounding the iron core. Such images are produced by mapping the phase angle shifts across the sample,18 which bring information about mechanical properties besides the topography features. As shown in Fig. 1b, AFM phase image allows the visualization of the soft NP coating (white areas) as well as of their stiffer magnetic core (darker areas).
2.2 Preparation of PNA decamers 1 and 2
To study the conjugation of the PNA strand onto SPION@DMSA, we selected, as model, a PNA decamer sequence containing all four nucleobases, namely GTAGATCACT. Since DMSA contains two different functional groups (–COOH and –SH), we synthesized the two PNA sequences 1 and 2 (Chart 2), displaying a terminal free amine group and a maleimido group, respectively, to verify the possibility of conjugation of PNA onto SPION through the amide bond formation reaction between PNA 1 and –COOH groups of DMSA, or through a Michael-type addition between PNA 2 and –SH groups of DMSA.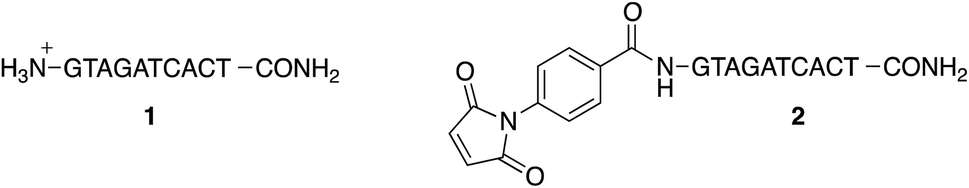 | ||
| Chart 2 The two PNA decamers here prepared. | ||
For this purpose, the resin-supported PNA decamer 3 (Scheme 2) was prepared by an automated solid-phase synthesis, using standard Boc/Z chemistry (see Experimental). The corresponding PNA decamer 1 was then obtained through the acidic treatment of Boc-3 (Scheme 2), which enables the cleavage of PNA 1 from the resin, removing at the same time all the Boc/Z protecting groups, followed by RP-HPLC purification of crude 1 (see Experimental). The preparation of PNA 2 was accomplished through the introduction of the maleimido moiety using p-maleimidobenzoic acid (4),19 which was manually coupled to the terminal NH2 group of the resin-supported PNA 3 by forming the amide bond with HATU as the condensing agent (eqn (b) in Scheme 2).
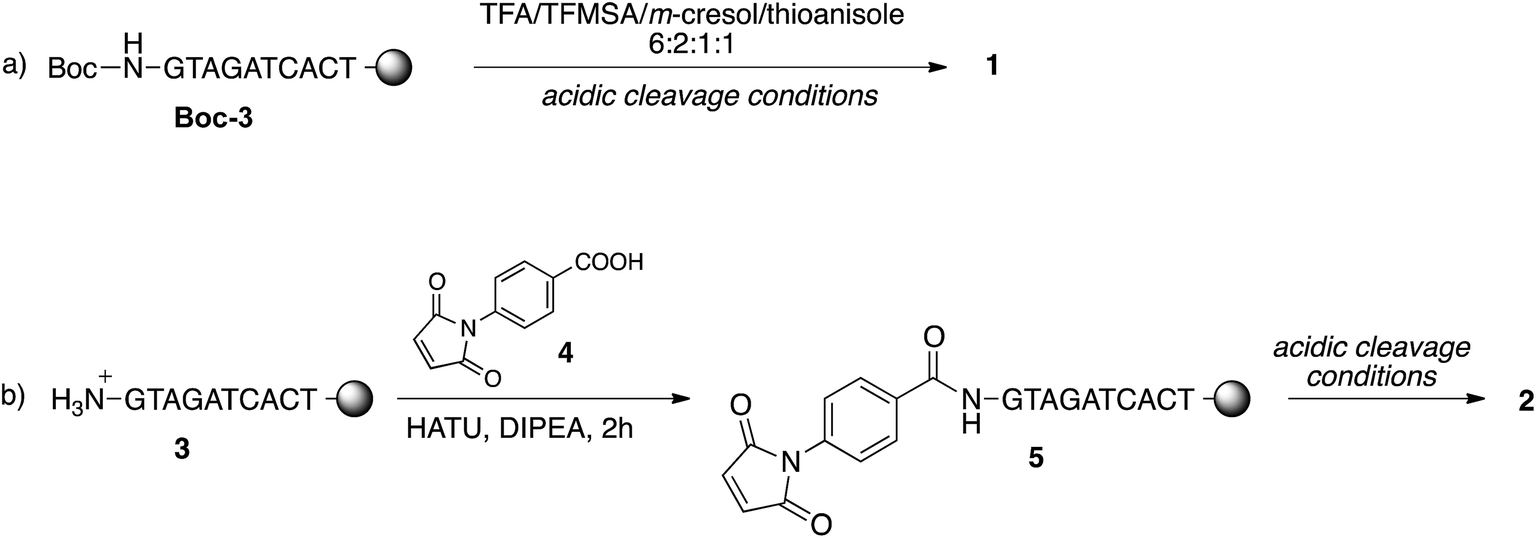 | ||
| Scheme 2 The synthesis of PNA decamers 1 and 2 from the resin-supported PNA decamers Boc-3 and 3. | ||
Thanks to the stability of the maleimido group toward the strong acidic conditions used for the cleavage of PNA from the resin, PNA 2 could be easily obtained from resin 5, and purified by RP-HPLC. The purity and identity of PNA 1 and 2 were confirmed by HPLC and MALDI-TOF analyses.
2.3 Conjugation of PNA to SPION
For the conjugation of the PNA strand to SPION, the two methods depicted in Scheme 3 were investigated. The widely used20 coupling reaction between COOH and NH2 groups, mediated by the zero-length cross linker EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride), was performed in the presence of NHS (N-hydroxysuccinimide), which gave the corresponding succinimidyl ester of the COOH groups, more stable in the aqueous environment then the corresponding isourea intermediate ester (bottom of Scheme 3). This link strategy has been previously used also for binding functional molecules (such as PEG,21 2-deoxyDglucose,22 or the NIRF dye IRDye800CW23) to the surface of NPs covered with DMSA. Unexpectedly, in our case this method did not work well. Some irreversible aggregation was observed (even in repeated trials) just a few minutes after the addition of EDC·HCl to the initially clear suspension of SPION@DMSA and NHS, which likely hampered the subsequent conjugation with PNA 1. In fact, the elemental analysis of the isolated final product clearly indicated that PNA was not the main species linked to the SPION surface, the measured C/N ratio being much higher (2.16, after subtraction of the carbon content due to DMSA, see Table 2) than expected for bound PNA (1.60).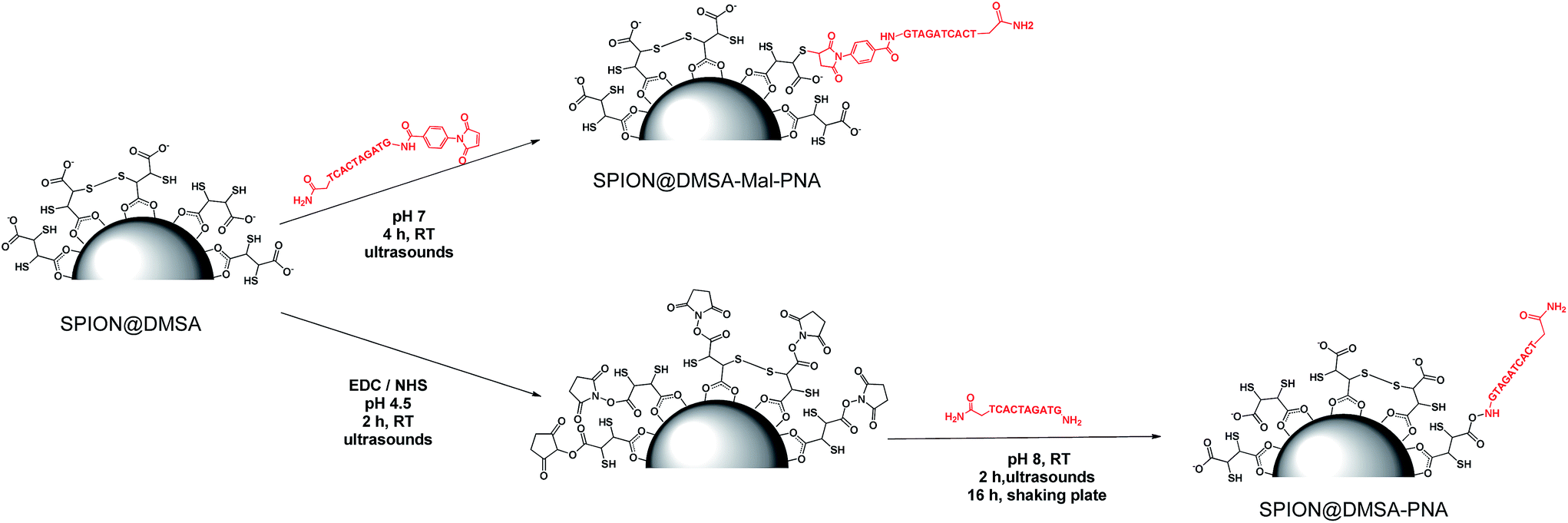 | ||
| Scheme 3 Schematic representation of the two investigated routes for binding PNA to SPION@DMSA. | ||
| SPION@DMSA-Mal-PNA | SPION@DMSA-PNAa | SPION@DMSA | PNA 1b | PNA 2b | |
|---|---|---|---|---|---|
| a Nanoparticles prepared by the amide bond formation between the NH2 of PNA and the COOH groups on SPION (see Scheme 3 for clarity).b Calculated values.c Calculated by subtracting the C% due to DMSA from the total C%. | |||||
| C% | 10.23 | 8.03 | 3.36 | 47.57 | 48.85 |
| H% | 1.40 | 1.73 | 0.75 | 5.03 | 4.86 |
| N% | 3.97 | 2.16 | n.d. | 29.79 | 28.25 |
| C/N ratio | 1.73c | 2.16c | — | 1.60 | 1.73 |
A different approach was therefore developed, involving the Michael addition of a thiol group of DMSA to the double bond of the maleimide-functionalized PNA 2 (top of Scheme 3). An Ellman's assay was preliminarily performed, to assure the presence of accessible SH groups on the SPION surface (see ESI†).
Moreover, before moving to the study of the conjugation of PNA 2 onto SPION@DMSA, the feasibility of the addition of SPION surface SH groups to a maleimide moiety was tested using a maleimide-rhodamine B adduct, expressly prepared by reacting 2-(aminoethyl)-maleimide with rhodamine-B isothiocyanate, as depicted in Scheme S1 (ESI†) and detailed in the Experimental.24 The reaction between the rhodamine-maleimide adduct and SPION@DMSA was conducted for 24 h at room temperature in the dark, to avoid the early photochemical decomposition of the chromophore. After accurate washing of the SPION (see Experimental), to remove all the unreacted chromophores, an UV-Vis spectrum of the suspension showed the superposition of the typical rhodamine peaks at 528 and 559 nm with the broad absorption/scattering profile of iron oxide SPION (Fig. S6, ESI†). The UV-Vis spectrum of the supernatant, measured after a further centrifugation cycle, showed very weak absorptions, due both to rhodamine and iron oxide SPION, attributable to the slight fraction of nanoparticles not removed in the centrifugation procedure. This indicated that the maleimide-rhodamine adduct had been really grafted to SPION, further confirming the presence on the nanoparticle surface of SH groups suitable for the Michael addition reaction.
On the basis of these results, the functionalized PNA 2 was reacted with SPION@DMSA, and the Michael addition was completed in 4 h at room temperature at neutral pH, under ultrasonic irradiation. The colloidal suspension, which remained clear throughout the coupling procedure, was centrifuged to isolate the functionalized nanoparticles (SPION@DMSA-Mal-PNA), while removing the unreacted PNA excess.
2.4 Characterization of SPION@DMSA-Mal-PNA
TEM images (Fig. 2) collected on the SPION@DMSA-Mal-PNA demonstrated that, as expected, the conjugation process did not affect neither the morphology nor the average size of the NP magnetic cores.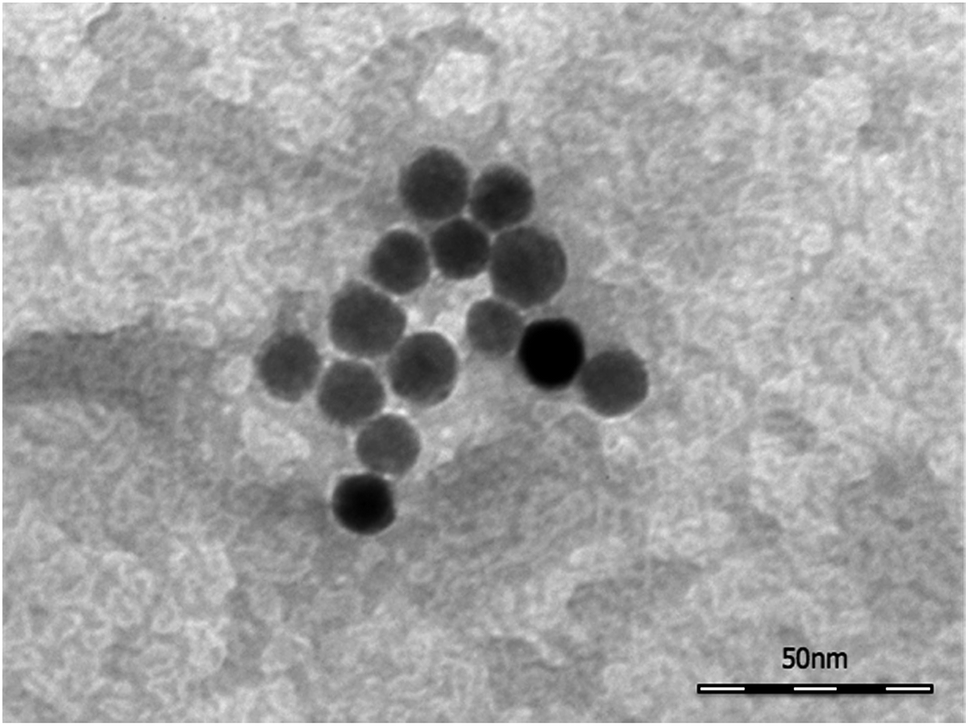 | ||
| Fig. 2 A representative bright field TEM image of SPION@DMSA-Mal-PNA. | ||
Moreover, DLS measurements showed the same size distribution with respect to the unfunctionalized SPION@DMSA (Fig S5, ESI†), proving that the conjugation of NPs to PNA did not affect the aggregation state.
The UV-Vis spectrum of SPION@DMSA-Mal-PNA suspended in water is reported in Fig. 3 together with that of the SPION@DMSA precursor. Their spectral difference (blue trace of the inset) clearly highlights the presence of the typical absorption peak of PNA nucleobases at 260 nm, slightly shifted with respect to the position (250 nm) for the free maleimide-PNA sequence (green trace in the inset), in line with previous evidence obtained on maghemite-PNA adducts.25
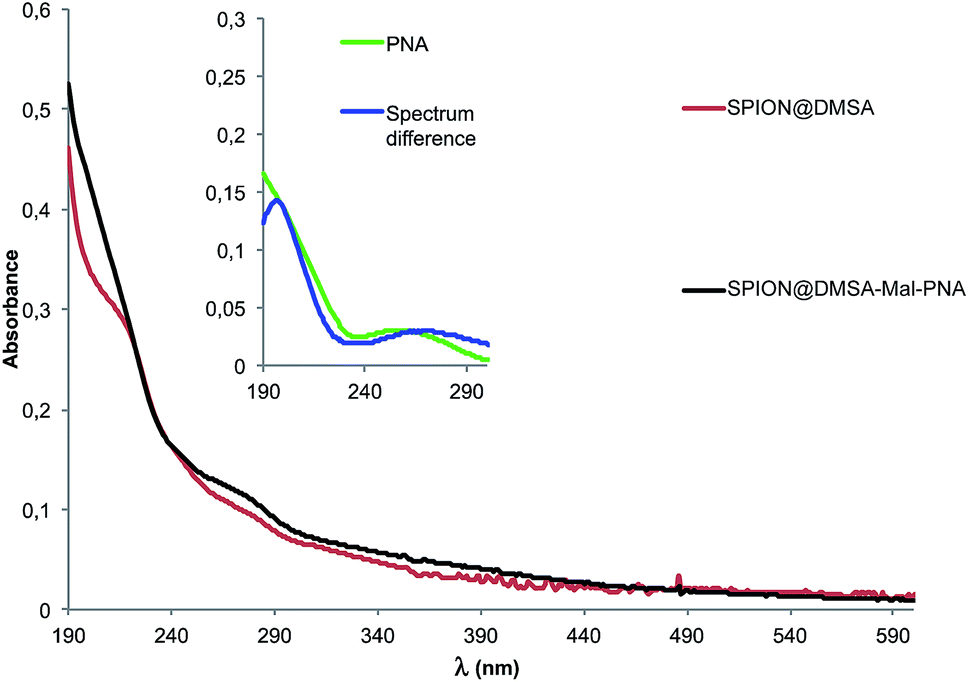 | ||
| Fig. 3 UV-Vis spectra of water suspended SPION@DMSA-Mal-PNA (black trace) and SPION@DMSA (red trace). Inset: blue trace, spectrum difference between SPION@DMSA-Mal-PNA and SPION@DMSA; green trace, spectrum of maleimide-PNA. | ||
The adducts have also been investigated by FTIR spectroscopy and Fig. 4 compares the spectrum of SPION@DMSA-Mal-PNA (lower trace) with that of SPION@DMSA (upper trace). In the latter trace, the absorption at 589 cm−1 is attributable to the Fe–O–Fe stretching modes of the spinel structure of magnetite,17,26 while the presence of DMSA is indicated by the weak absorptions at about 2900 cm−1 (due to aliphatic νCH modes), and by the broad bands at 1632 and 1396 cm−1, which are related to the asymmetric and symmetric stretching of the COO− groups of DMSA.27 The strong and broad νOH band at ca. 3400 cm−1 indicates the presence of water on the nanoparticle surface, and therefore the absorption at 1632 cm−1 also includes contribution from the OH bending modes. The main difference between the two traces of Fig. 4 is constituted by the presence in the lower trace of a series of relatively weak absorptions in the 1100–1400 cm−1 region, due to the C–N stretching modes of the nucleobases and of the PNA amide skeleton.28 The nucleobases and the amide CO groups of PNA also contribute to the broad absorption at ca. 1660 cm−1, which is more intense and slightly shifted with respect to the spectrum of the SPION@DMSA precursor.
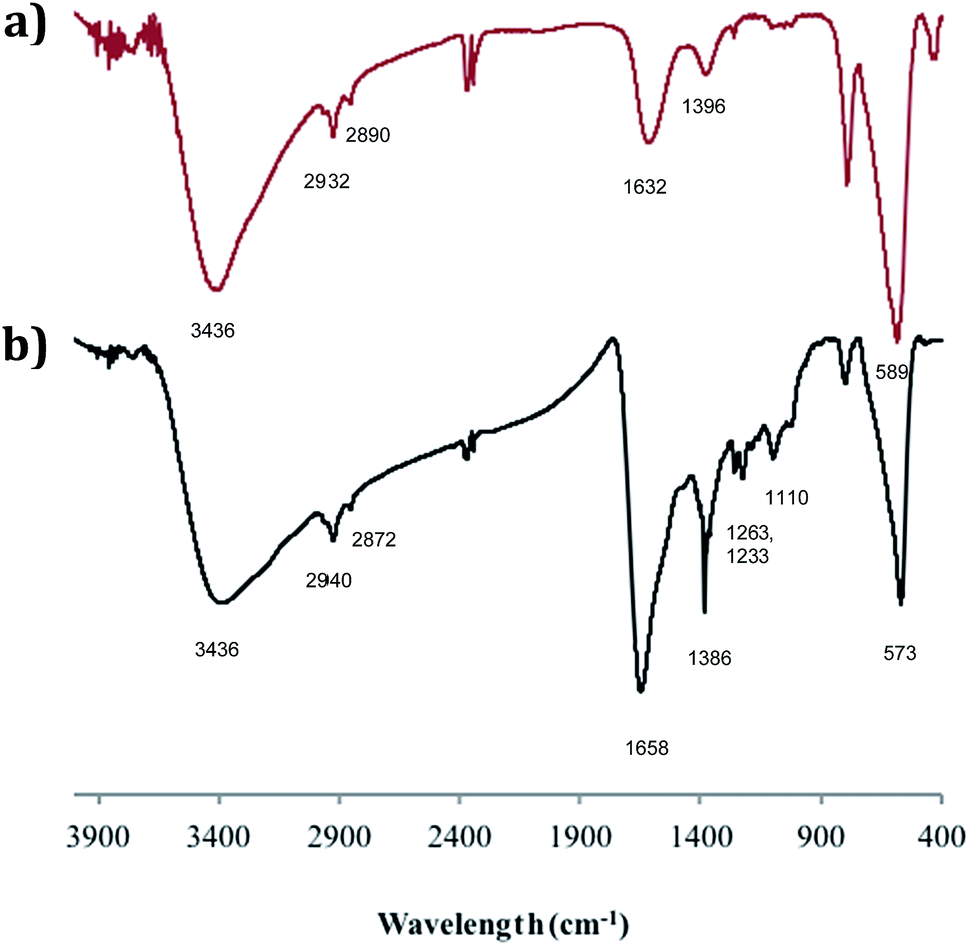 | ||
| Fig. 4 FTIR spectra (KBr pellet) of (a) SPION@DMSA; (b) SPION@DMSA-Mal-PNA. | ||
To prove the effectiveness of the addition of PNA 2, elemental analyses were performed on the dried nanoparticles. Data are reported in Table 2.
The analyses showed the expected significant increase of the C and N content after interaction with PNA (the H% values were not considered, since the use of water as solvent affects the reliability of these data). Noteworthy, the C/N ratio for the SPION@DMSA-Mal-PNA adduct (after subtraction of the DMSA contribution) was in perfect accord with the calculated value for the Mal-PNA strand here used. On these bases, the PNA loading onto the SPION@Mal-PNA was calculated to be about 14% w/w, corresponding to a molar ratio of ca. 1:15 with respect to DMSA. Each SPION@DMSA-Mal-PNA nanoparticle, which is covered by ca. 6000 DMSA molecules, is therefore loaded with ca. 400 PNA strands.
On the contrary, the C/N ratio for the nanoparticles SPION@DMSA-PNA prepared from PNA 1 (Scheme 3) was much higher than the one of PNA, suggesting that, as above discussed, the sample was largely contaminated by impurities with a higher C/N ratio, likely deriving from reagents (EDC, NHS) used in the coupling procedure and only partially removed.
Finally, ζ-potential measurements on the SPION@DMSA-Mal-PNA colloid (Fig. 5) showed a decrease of the negative surface charge with respect to SPION@DMSA, from −44.5 ± 1.0 mV to −27.7 ± 4.0 mV, in line with the bonding of a positively charged species such as PNA to the negatively charged native nanoparticles.
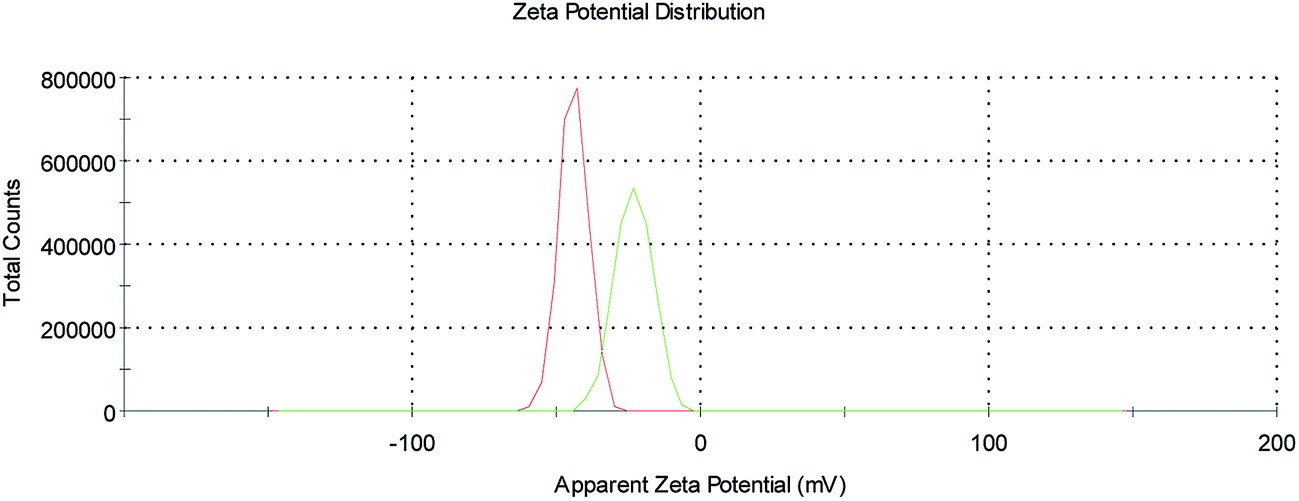 | ||
| Fig. 5 ζ-Potential profiles of SPION@DMSA (red trace) and SPION@DMSA-Mal-PNA (green trace) in water, at pH 7.4. | ||
2.5 Magnetic and relaxivity properties of SPION
Thermal energy released by SPION during the action of an alternating magnetic field is the crucial parameter for the success of the MFH treatment. The capability of releasing heat is normally quantified by the SAR (Specific Absorption Rate), which describes the absorbed power per mass unit. In the linear regime, i.e. when the magnetization varies linearly with the magnetic field (low field amplitude and low energy barrier compared to thermal energy) the SAR depends on the field parameters and on the intrinsic properties of the SPION (eqn (1)),29a where ν and H0 are the frequency and amplitude of the oscillating field, MS is the saturation magnetization, T the temperature, kB the Boltzmann constant, ρ the mass density, V the particle volume and τ the time required by the particle's magnetic moment to reverse its orientation.
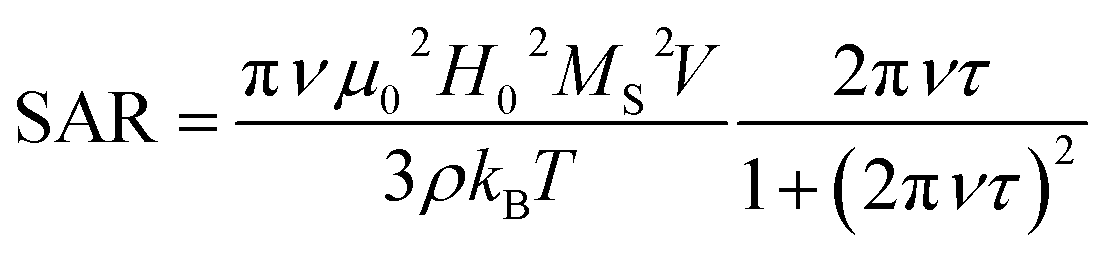 | (1) |
Thus, to maximize the SAR, SPION must exhibit specific magnetic characteristics.29 Indeed, despite the SAR can be also increased by using high frequencies and high magnetic fields, these parameters cannot be increased above a fixed threshold, due to severe limitation imposed by the biological systems.30 On one hand, the SAR reaches its maximum value when the reversal time equals the time of the magnetization corresponding to the working frequency ν = 183 kHz, i.e. τ = 1/(2πν). For magnetite NPs this condition is satisfied for size in the 16–19 nm range31 as is the case of SPION7. On the other hand, larger SAR will be obtained with higher saturation magnetization, which therefore should be as close as possible to the bulk magnetite value (92 emu g−1).32
Magnetic measurements were performed on powder samples of SPION7, (which is here indicated simply as SPION, as stated at point 2.1 above) both before and after the ligand exchange, using a SQUID magnetometer. The main magnetic parameters of the measured samples are listed in Table 3.
| TB (K) | HC (Oe) | MS (emu g−1) | M0 T/M5 T | |
|---|---|---|---|---|
| SPION@OA | 250 | 460 | 87 | 0.30 |
| SPION@DMSA | 270 | 430 | 82 | 0.36 |
| SPION@DMSA-Mal-PNA | 270 | 430 | 80 | 0.36 |
As shown in Fig. 6a the zero field cooled–field cooled (ZFC–FC) magnetizations of SPION@OA display the characteristic blocking process of single domain NPs. The FC magnetizations exhibit a plateau at low temperature, which can be ascribed to interparticle interactions, which are not negligible when NPs are measured as dried powders. The blocking temperature, estimated as a first approximation from the maximum of the ZFC curve, is TB = 250 K, indicating that at room temperature the NPs are in the superparamagnetic regime, which is a fundamental requisite for the proposed applications. This result is confirmed by the absence of remanence (Fig. 6b) observed in the room temperature magnetization vs. field curve. Conversely, at low temperature (Fig. 6c) magnetic irreversibility is observed, with a coercive field HC = 500 Oe and reduced remanence R = MR/MS = 0.3, comparable with the data reported in the literature for magnetite nanoparticles of similar size.33 Most importantly, the saturation magnetization is high (MS = 85 emu g−1) and comparable to the value of bulk magnetite. A fit to the Langevin function of the low field part of the M(H) curve34 gives a magnetic moment value per particle, μ, of ca. 5 × 104 μB, which is consistent with the size and magnetization of the NPs, confirming their good quality.
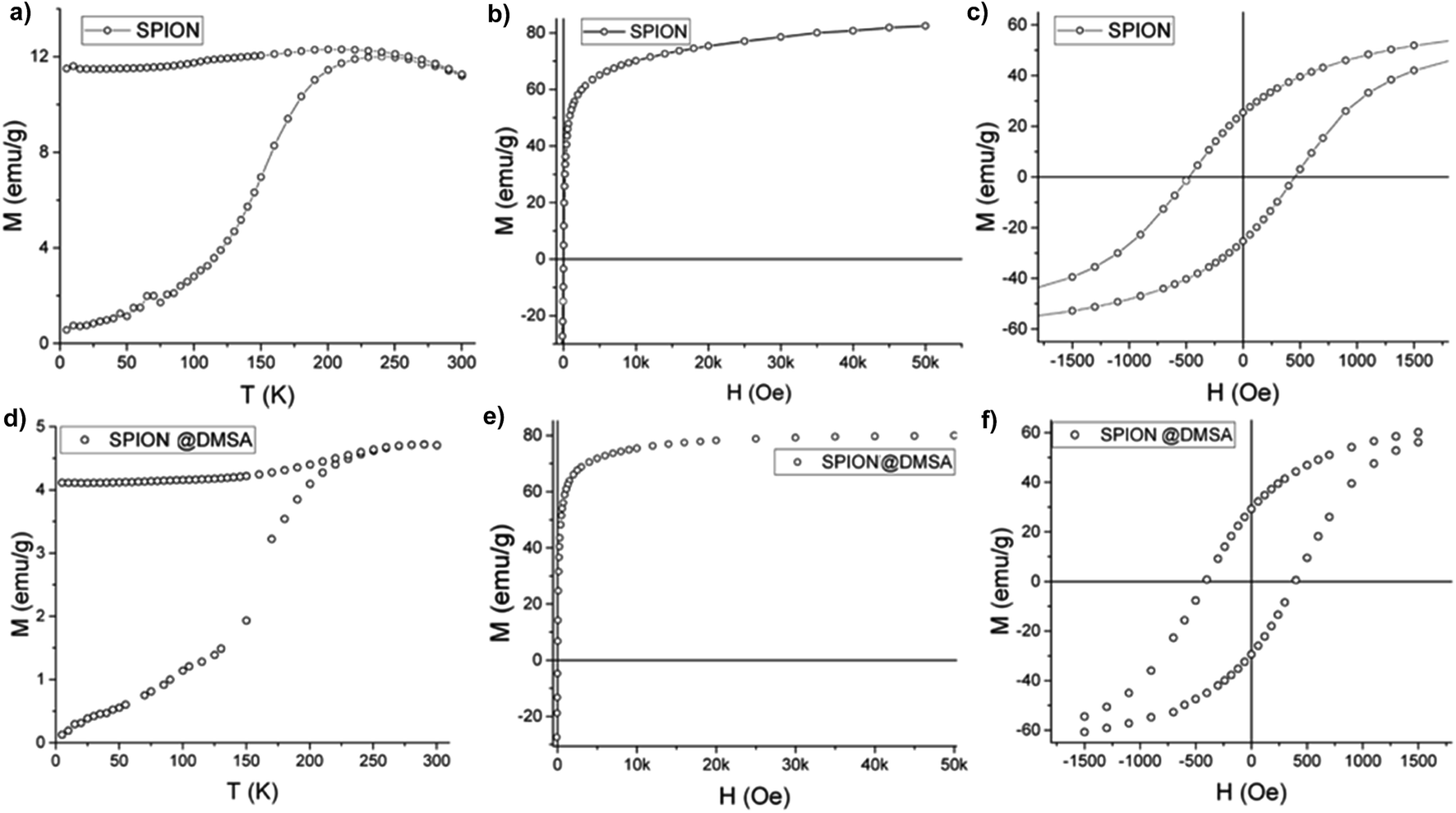 | ||
| Fig. 6 Magnetic data on SPION@OA (upper panels) and SPION@DMSA (lower panels): (a and d) ZFC-FC magnetization curves; (b and e) magnetization at 300 K; (c and f) enlargement of the low field region of the hysteresis cycle at 2.5 K. | ||
The magnetic data of SPION@DMSA reported in Fig. 6d–f show that the ligand exchange does not affect the good magnetic characteristics: TB lower than room temperature, no remanence at 300 K and high MS value. The same holds true for the NPs after their conjugation to PNA (Fig. S7, ESI†).
Magnetic hyperthermia measurements were made by recording the evolution of temperature of the SPION@DMSA samples dispersed in water (0.77 mg mL−1 Fe) exposed to an alternating magnetic field of 17 kA m−1 and 183 kHz for 5 minutes. The temperature kinetics is shown in Fig. 7. Despite the small temperature increase observed, the sample has a remarkable SAR = 65 W g−1 Fe, which makes it a suitable candidate for future application in MFH treatments.
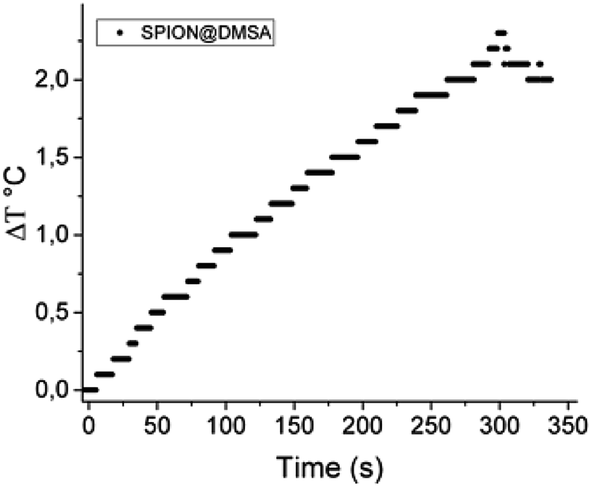 | ||
| Fig. 7 Temperature kinetics of a water suspension of SPION@DMSA, 0.77 mg mL−1 Fe, during the exposure to the alternating magnetic field (17.1 kA m−1, 183 kHz). The starting temperature was 295 K. | ||
The ability of SPION@DMSA to affect water nuclear relaxation times was also measured, to evaluate their effectiveness as MRI contrast agents. In Fig. 8 the 1H-NMR dispersion (NMRD) profiles for SPION@DMSA as well as for SPION@DMSA-Mal-PNA are reported, where the nuclear longitudinal (r1) and transverse (r2) relaxivities were evaluated in the usual way (eqn (2)), in which (1/Ti)meas indicates the measured value on the sample with iron concentration c = 0.5 mmol L−1, and (1/Ti)dia the nuclear relaxation rate of the water used as host solution.
| ri = [(1/Ti)meas − (1/Ti)dia]/c, i = 1, 2 | (2) |
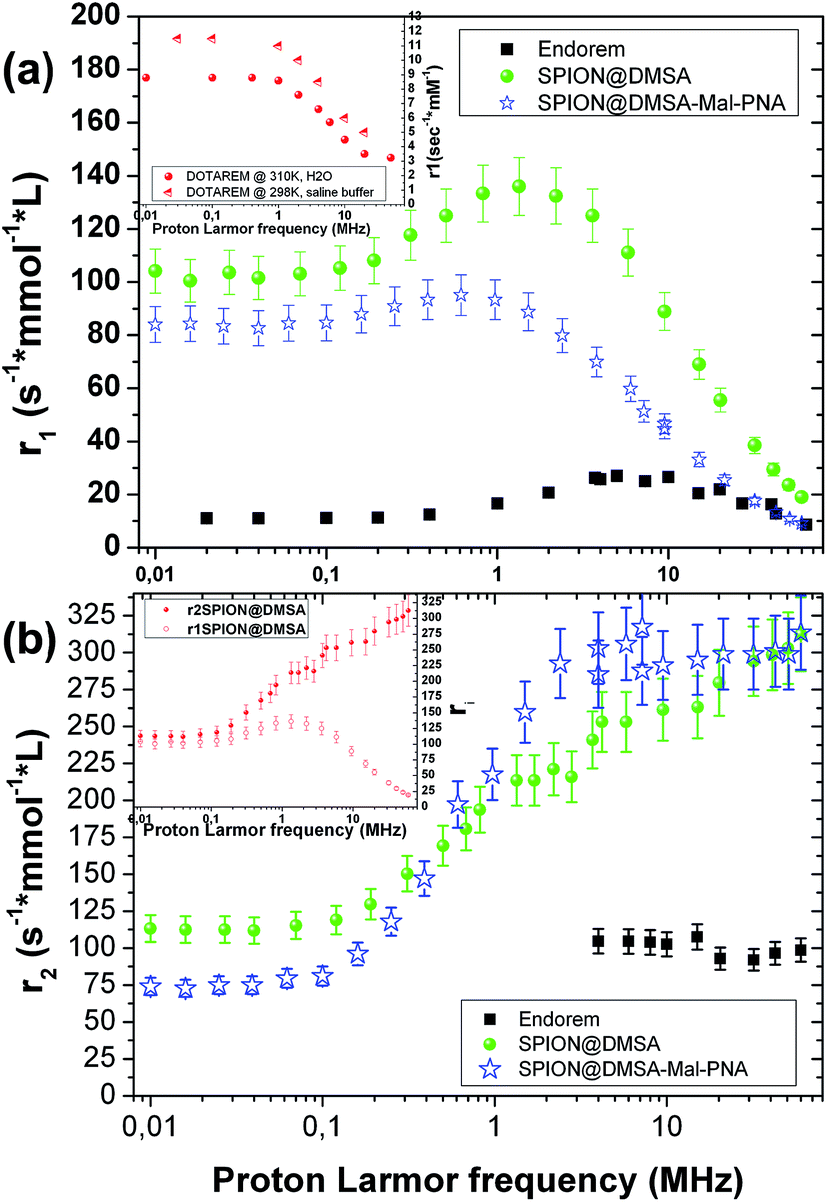 | ||
| Fig. 8 Longitudinal (a) and transverse (b) NMRD profiles of SPION@DMSA (green circles) and SPION@DMSA-Mal-PNA (blue stars) compared to commercial compound Endorem® (black squares). In the inset of panel a, the longitudinal NMRD profiles of the commercial contrast agent Dotarem dispersed in water or in saline buffer measured at two different temperatures are reported. The inset shows the extremely good performances of both our samples also as T1 relaxing agent. In the inset of the panel b, longitudinal and transverse relaxivities of SPION@DMSA are plotted together (the same results are obtained also for SPION@DMSA-Mal-PNA), to show the equivalence of r1 and r2 values at low frequency, as predicted by the theory (see the text). | ||
It can be evinced that the sample is able to effectively shorten both longitudinal (T1) and transverse (T2) relaxation times, and therefore it strongly enhances the correlated relaxivities.
In particular, the r2 (the crucial parameter for a superparamagnetic iron oxide material) is 2 or 3 times higher than the corresponding value of the commercial contrast agent Endorem® at the typical frequencies of the clinical instrumentation [ca. 8.5, 21.3 and 63.9 MHz (magnetic fields of 0.2, 0.5 and 1.5 T, respectively)]. Furthermore SPION@DMSA possess better r1 values (more than one order of magnitude) with respect to the routinely used positive contrast agent Dotarem® (see inset in Fig. 8a) at those frequencies/fields. This property opens the possibility to use SPION@DMSA as positive and negative contrast agent at the same time, depending on the experimental parameters used in the pulse NMR sequences during the acquisition of MR images.35 Interestingly, SPION@DMSA shows no difference between r1 and r2 at the zero field limit (see inset in Fig. 8b) as predicted by theory, an experimental result obtained only recently in the literature.36 The equivalence of r1 and r2 at low proton Larmor frequencies would have been unattainable if our sample was not well dispersed in water without any unwanted particle aggregation.
The slight influence of the functionalization with PNA on the relaxivity values is explained by two characteristics of SPION@DMSA-Mal-PNA: on one side the higher steric hindrance as evinced by AFM (NP overall size = 25.6 ± 2.6 nm (averaged value over 38 observations)), and on the other side the poor solubility of PNA sequence. Both these properties oblige the water molecules to stay slightly further from the magnetic core of the SPIONs or, at least, to follow a more difficult pathway if compared to the case of SPION@DMSA. Consequently, mildly lower values for the r1 NMR dispersion profile and for low proton Larmor frequencies of r2 are found, while substantially the same r2 values of SPION@DMSA sample are obtained at ν > 0.1 MHz.
Therefore, the PNA presence did not influence significantly the relaxivity efficiencies of these nanosystems, whereas the functionalization provides a significant added value.
3. Conclusions
A novel and effective strategy for binding PNA oligomers onto the surface of superparamagnetic iron oxide nanoparticles has been developed. All the requirements that stimulated this work have been fulfilled, and some of the drawbacks shown by previous literature reports have been overcome. The control of the size of the magnetic core for the diagnostic and therapeutic applications is ensured by the use of thermal decomposition synthetic route. The desired water solubility and the colloidal stability are provided by the coating with DMSA. The stable linking of SPION to PNA oligomers is given by the efficient Michael addition of maleimide-functionalized PNAs to the SH groups of DMSA grafted to the nanoparticle surface.A detailed characterization of the morphological, magnetic and relaxivity properties of the SPION, which have been further conjugated to PNA, showed that these nanoparticles are well-suited to increase both the positive and negative contrast of magnetic resonance imaging and to rise the temperature of their surroundings when subjected to an alternating magnetic field (with a SAR as high as 65 W g−1). Noteworthy, the conjugation of PNA to NPs did not affect their magnetic and relaxivity properties.
The approach here described, therefore, produces multimodal hybrid organic–inorganic nanomaterials, that can act as dual (T1 and T2) MRI contrast agents, as efficient hyperthermia promoters and as PNA carriers. To fully exploit the theranostic potential of these nanohybrids, future work will explore the replacement of the standard PNA sequence, here used for setting the method, with PNA oligomers designed for targeting specific non-coding microRNAs (miRNA),37 whose dysregulation has been implicated in a variety of pathologies, such as inflammatory and autoimmune diseases, neurological disorders, as well as several types of cancer.38
4. Experimental part
4.1 Materials
All the reagents have been purchased by Sigma Aldrich (reagent grade) and used without further purification, if not otherwise specified. The PNA decamers were synthesized with standard automated Boc-based chemistry using commercially available monomers purchased from ASM Research Chemicals (Burgwedel, Germany). Polystyrene bead carrying 4-methylbenzhydrylamine hydrochloride salt groups (MBHA resin, 0.63 mmol g−1) was purchased from VWR International, and it was downloaded to 0.2 mmol g−1 with the thymine monomer Boc-PNA-T-OH as described elsewhere.254.2 Instruments and methods
Automated solid phase synthesis of resin-supported PNA 3 was performed with peptide synthesizer ABI 433A of Applera Italia, according to Applied Biosystems ABI 433A Peptide Synthesis 3 mL Reaction Vessel User's Manual for the MBHA resin. The software for peptide synthesis was Synassist 2.0.Mass spectra were recorded on a ESI-Q-Tof Micro-Waters (Waters Corporation, Milford, MA), in the data-dependent acquisition and positive ion mode.
The reverse-phase RP-HPLC analyses were performed on Agilent 1100 series system, equipped with DAD analyser. The PNA purification was performed by RP-HPLC with UV detection at λ 260 nm using the semi-preparative column Luna C18 (25 cm × 10 mm, 5 μm), eluting with H2O containing 0.1% TFA (eluent A) and CH3CN containing 0.1% TFA (eluent B); elution gradient: from 90% A to 50% B over 30 min; flow rate: 3 mL min−1. The purity of PNA was evaluated by RP-HPLC using analytical column Luna C18 (25 cm × 4.6 mm, 5 μm), eluting with H2O containing 0.1% TFA (eluent A) and CH3CN containing 0.1% TFA (eluent B); elution gradient: from 95% A to 100% B over 60 min; flow rate: 1 mL min−1.
High resolution NMR experiments were performed on a Bruker Model DRX400 spectrometer, equipped with a Bruker 5 mm BBI Z-gradient probe head capable of producing gradients with a strength of 53.5 G cm−1.
Infrared (IR) spectra were acquired on a Perkin-Elmer Spectrum BX FT-IR instrument. Samples were dispersed in KBr and pressed in a pellet. The IR spectra were registered between 4000 and 400 cm−1.
Electronic absorption spectra were recorded on an Agilent Model 8543 spectrophotometer at room temperature and using quartz cells with 1.0 cm path length.
Metal content on SPION was determined by AAS analysis on a Perkin-Elmer Pinaacle 900 instrument and for SPION7@DMSA was further confirmed by a spectrophotometric method. For AAS analysis few microliters of particle suspension were digested with 1 mL aqua regia/HCl overnight at RT in a 10 mL volumetric flask, and subsequently filled up with milliQ water. For spectrophotometric analysis few microliters of particle suspension were digested with 1 mL aqua regia/HCl at RT in a 10 mL volumetric flask and then filled up with (i) NH2OH solution (10% w/w, 0.1 mL), (ii) acetate buffer solution (pH 4.6, 0.15 M, 6 mL), (iii) 1,10-phenantroline solution (0.6% w/w, H2O/MeOH 10:1, 0.2 mL) (iv) milliQ water. The absorbance at 510 nm was then measured with an UV-Vis spectrophotometer and plotted against standards prepared with the same procedure starting from a commercial AAS standard solution.
Elemental C, H, N analyses were performed on a Perkin Elmer CHN 2400 instrument.
DLS and ζ-potential measurements were carried out on a Zetasizer Nano ZS instrument (Malvern Instruments Corp., Malvern, Worcestershire, UK) at a wavelength of 633 nm with a solid state He–Ne laser at a scattering angle of 173°, at 298 K on diluted samples (0.01–0.1 mg mL−1 nanoparticles) at pH 7. Each hydrodynamic diameter as well as ζ-potential were averaged from at least three measurements.
Average diameter and size distribution of the SPION were determined from TEM images recorded using a CM12 PHILIPS transmission electron microscope operating at 100 kV. Samples were prepared by drop drying a dilute solution of the samples onto 200 mesh carbon-coated copper grids. The recorded micrographs were further analyzed with the FIJI® software. The mean diameter and size distribution of each sample were obtained from a statistical analysis over 400 SPION.
The NMR-dispersion profiles were measured on water suspensions of each sample. Longitudinal and transverse nuclear relaxation times, T1 and T2, respectively, were evaluated over the range 10 kHz to 60 MHz for the 1H (proton) Larmor frequency, corresponding to an applied magnetic field in the range 2.3 × 10−4 to 1.41 T. In order to cover such a wide range, two different instruments were used: a Stelar Spinmaster with a standard electromagnet for the range: 10–60 MHz; and a Stelar SMARtracer, working with the Fast-Field Cycling technology, for the low and very low fields' range, corresponding to 10 kHz to 10 MHz. In the first case, standard pulse sequences were selected, that is, saturation recovery for T1 and Car–Purcell–Meiboom–Gill (CPMG) for T2. For the very low field range (υ < 3.7 MHz for T1 and υ < 4:2 MHz for T2), ad hoc pre-polarized sequences were used to increase the NMR proton signal.
AFM imaging was performed using a Nanoscope Multimode IIId AFM (Bruker, Santa Barbara, CA, USA). Tapping-mode AFM images were collected in air using the root mean square amplitude of the cantilever as the feedback signal for the vertical sample position. The mica support (Ted Pella, CA, USA) was glued to a metal disk that was magnetically fixed to the AFM sample holder. Rectangular silicon nitride probes with nominal spring constant around 2.5 N m−1 (NSG01, NT-MDT, Russia) and cantilever length of 120 μm were used for the tapping-mode imaging. The cantilever resonance frequency was about 130 kHz. The root mean square free amplitude of the cantilever was approximately 15 nm and the relative set-point above 95% of the free amplitude. Images were recorded at ∼1 Hz line rate, and a resolution of 512 × 512 pixels per image was chosen. AFM images were subjected to a line-by-line subtraction of linear background (“flattening”), to eliminate sample tilt from the images and correct for stepwise changes between individual scan lines by using the Nano-Scope III software (version S.31R1, Bruker, USA).
Magnetic measurements were performed using a Quantum Design MPMS SQUID magnetometer operating in the 1.8–350 K temperature range and with an applied field up to 50 kOe. Measurements were performed on pressed powder of SPION sample. All data were corrected for the diamagnetic contribution of the sample holder, and were normalized to the amount of magnetic material as evaluated from elemental analysis. Zero Field Cooled–Field Cooled (ZFC/FC) curves were obtained by measuring the temperature dependence of the magnetization applying a probe magnetic field (50 Oe), after cooling the sample in the absence (ZFC) or in the presence (FC) of the field. A rough estimate of TB value accuracy is given by the half interval between the temperatures of two subsequent acquisition points, i.e., 2.5 K. The field dependence of the magnetic moment (M vs. H) was measured cycling the field between ±50 kOe at 2.5 K and 300 K. The accuracy of HC, MR and MS evaluation can be estimated as low as 3%.
The determination of Specific Absorption Rate (SAR) was performed through calorimetric measurements by recording temperature kinetics of SPION suspension exposed to an alternating magnetic field. Measurements were performed adapting a commercial setup, composed by a 6 kW Fives Celes power supply, a water-cooled induction coil and a series of variable capacitors (420 nF to 4.8 μF) for setting the required frequency. Such resonant RLC circuit is able to produce an alternating magnetic field in the range of 50–400 kHz and with amplitude up to 19.1 kA m−1. The frequency and field amplitude values used in this work (183 kHz, 17.0 kA m−1) were chosen in order to operate under the physiological limit, Hν < 5 × 109 A m−1 s−1, beyond which deleterious responses of living tissues are observed. The sample was placed in the middle of the induction coil, inside a polystyrene sample holder placed in a glass Dewar connected to a glycol thermal bath in order to isolate the sample bath from the thermal gradient of the coils and from the environment. The real amplitude of the magnetic field was determined by a AMF Life Systems high frequency probe. Measurements of the sample temperature were performed by an optical fiber thermometer connected to a digital temperature recorder (Fotemp). The SAR values were calculated using the equation SAR = (Σimici/mme)(ΔT/Δt), where ΔT is the temperature increase in the interval of time Δt, mme is the total mass of metal, mi is the mass in grams of the i-species and ci is its specific heat. The sum is extended to all the i species involved in the heat exchange. Since the measurements are carried in non adiabatic conditions, the ΔT/Δt values were extrapolated for t ≈ 0 from temperature kinetic curves, by considering the initial slope.
4.3 Synthesis of SPION7@OA
The synthesis of SPION was performed following a slightly modified literature procedure.11 Briefly, in a 50 mL three-necked round bottom flask 13.9 mmol of oleic acid (4.38 mL, d = 0.895 g mL−1; 70% purity) and 2 mmol of Fe(CO)5 (236 μL, d = 1.49 g mL−1) were dissolved under inert atmosphere in 4 mL of 1-octadecene. This solution was then heated from room to 320 °C at a rate of 15 °C min−1 and refluxed for 3 h. During this time the reddish solution turned to black. The solution was then cooled to 120 °C at a rate of 5 °C min−1 and maintained at that temperature for 2 more hours in air to allow oxidation of Fe(0). The solution was cooled down to room temperature at a rate of 5 °C min−1. The SPION were subsequently isolated by centrifugation of the reaction mixture after addition of ca. 70 mL of acetone (3× at 7197 round centrifugal force, rcf) followed by removal of the yellow supernatant. The isolated particles were then re-suspended in 15 mL of hexane and stored under nitrogen atmosphere at −25 °C for further uses.The same procedure was followed for the synthesis of SPION6@OA and SPION8@OA, using different amounts of oleic acid (1.90 mL and 4.9 mL, respectively), corresponding to 6 and 8 equivalents, with respect to Fe(CO)5.
4.4 SPION@DMSA
A sample of SPION7@OA in hexane solution (3 mL, 11 mg mL−1 Fe as measured by AAS) was treated with 8 mL of acetone and centrifuged (4 min, 7197 rcf) to remove the excess oleic acid. The nanoparticles were then suspended in 9 mL of hexane and moved to a Schlenk flask under nitrogen atmosphere. To this suspension a solution of DMSA dissolved in acetone (43.5 mg in 9 mL) was added, followed by 15 μL of TEA. The Schlenk flask was then moved to an ultrasonic bath and the reaction mixture was kept at 50 °C for 40 min, maintaining the suspension under nitrogen atmosphere for the whole sonication time. The SPION were then collected with a magnet and then centrifuged first with 20 mL of acetone (10 min, 7197 rcf) and then with 20 mL of water (30 min, 7197 rcf). The washed SPION were then re-suspended in 10 mL of water and stored under nitrogen atmosphere at 4 °C for further uses.4.5 Preparation of the resin-supported PNA decamer 3
Automated solid phase synthesis was performed on an ABI 433A peptide synthesizer in a reactor of 3 mL on a 20 μM scale using Boc strategy. The MBHA resin downloaded with thymine monomer (100 mg, 0.2 mmol g−1) was swollen with CH2Cl2, the Boc group of the loaded monomer was removed by treatment with TFA/m-cresol (95:5), the resin was rinsed with CH2Cl2 and DIPEA 1.6 M in NMP. The monomers were loaded into cartridges as NMP solutions, activated with HBTU 0.38 M in NMP and transferred to the resin. After each coupling step (30 min) the resin was washed with NMP and then treated with Ac2O/Py/NMP 1:25:25 capping solution twice for 3 min. The cycle was repeated for each base. After the last coupling the resin was washed several times with NMP and CH2Cl2, and finally dried under nitrogen yielding 3 (128 mg).
4.6 Synthesis of PNA decamer 1
50 mg of resin was washed with TFA (2 × 200 μL) and then stirred for 1 h with a solution of TFA/TFMSA/thioanisole/m-cresol 6:2:1:1 (500 μL). The mixture was filtered and the resin washed with TFA (4 × 200 μL). The filtrate was concentrated, and Et2O (5 mL) was added to precipitate PNA as a white solid. Centrifugation of the slurry gave the product, which was washed with Et2O (3 × 5 mL), and dried. The crude product was purified by RP-HPLC to afford the decamer 1 as a colourless solid (15 mg). Analytical RP-HPLC: tR = 8.86 min. ESI MS: m/z found 1363.9 [MH2]+, 909.6 [MH3]+, 682.4 [MH4]+, 546.2 [MH5]+; Mr calcd: 2726.6.
4.7 Synthesis of PNA decamer 2
The resin 3 (50 mg, 0.2 mmol g−1) was swollen with CH2Cl2 (3 mL) for 1 h, then the Boc group of the last monomer of the sequence was removed by treatment with a solution of TFA/m-cresol (95:5). The resin was rinsed with CH2Cl2 and NMP. In a vial, a solution of DIPEA (17 μL, 0.1 mmol, 10 eq.) and the p-maleimidobenzoic acid (11.9 mg, 0.055 mmol, 5.5 eq.) in NMP (0.4 mL) was added to a solution of HATU (19.0 mg, 0.05 mmol, 5 eq.) in NMP, and the resulting mixture was shaken for two minutes. The activated mixture was then added to the resin and shaken for 2 h. The resin was then washed with TFA (2 × 200 μL), and subsequently stirred for 1 h with a solution of TFA/TFMSA/thioanisole/m-cresol 6:2:1:1 (500 μL). The mixture was filtered, and the resin washed with TFA (4 × 200 μL). The filtrate was concentrated, and Et2O was added to precipitate PNA as a white solid. Centrifugation of the slurry gave the product, which was washed with Et2O (3 × 5 mL), and dried. The crude PNA was purified by RP-HPLC to afford the PNA 2 (11 mg) as a white solid. Analytical RP-HPLC: tR = 10.2 min. ESI MS: m/z found 1463.6 [MH2]+, 976.1 [MH3]+, 732.3 [MH4]+, 585.8 [MH5]+; Mr calcd: 2925.3.
4.8 Preparation of SPION@DMSA-PNA
The SPION@DMSA carboxyl groups were first activated by adding 12.4 mg EDC (0.065 mmol) and 46 mg NHS (0.4 mmol) to 5 mL of SPION@DMSA water suspension (0.56 mg mL−1 Fe). The addition of EDC apparently destabilized the nanoparticle colloid, resulting in visible aggregation. The suspension pH was then changed to 4.5 by careful addition of HCl and the activation reaction continued for 2 h in an ultrasonic bath. Then the activated SPION were added dropwise to a solution of 5 mg PNA 1 dissolved in 0.5 mL of CH3CN and 1 mL of phosphate buffer (pH 8, 5 mM). The suspension pH was adjusted to 8 by careful addition of NaOH solution. The reaction mixture was first placed in an ultrasonic bath and left under ultrasonic irradiation for 2 h at room temperature and then left on a shaking plate overnight. The nanoparticles were then purified via centrifugation with water (3×, 15 min, 7197 rcf) and subsequently collected in a Schlenk flask under inert atmosphere.4.9 Ellman's essay
An aliquot of 200 μL of SPION@DMSA suspension (0.56 mg mL−1 Fe) was diluted with 3 mL of phosphate buffer (pH 8.3, 0.05 M). To this suspension 1 mL of Ellman's reagent solution (3 mg dissolved in 10 mL phosphate buffer) was added. As a comparison two solutions, containing only SPIONs and only Ellman's reagent, respectively, were prepared. After 90 min of incubation in the dark the suspensions were centrifuged and the supernatant absorbance at 412 nm was measured by UV-Vis spectroscopy (Fig. S8, ESI†).4.10 Synthesis of the rhodamine B-maleimide adduct
The synthesis (depicted in Scheme S1, ESI†) involved at first the preparation of mono-protected N-Boc-ethylenediamine, that was performed according to a literature procedure.39 N-(2-Aminoethyl)maleimide was then prepared by another literature method,40 and reacted with rhodamine B isothiocyanate, in an NMR tube under nitrogen atmosphere: 3.3 mg of rhodamine B-NCS (6.15 × 10−3 mmol) were dissolved in 0.5 mL of deuterated methanol and treated with 1.6 mg of N-(2-aminoethyl)maleimide trifluoroacetate salt (5.12 × 10−3 mmol) and 0.85 μL of Et3N. The reaction was carried out overnight at RT and its progress was monitored following the decrease of the sharp singlet at 6.91 ppm due to the CH on double bond of N-(2-aminoethyl)maleimide that, once reacted, slightly shifted to upper fields (ca. 6.89 ppm) and split in multiple singlets due to the different isomers. The concentration of rhodamine-B-maleimide adduct was estimated to be 0.006 M using its known molar attenuation coefficient (ε = 87000 at 559 nm).41
4.11 Reaction between rhodamine B-maleimide adduct and SPION@DMSA
In a Schlenk flask under nitrogen atmosphere 100 μL of SPION@DMSA (0.78 mg mL−1 Fe) were diluted with 2 mL milliQ water. To this suspension 17 μL of the functionalized rhodamine solution were added. The reaction was incubated in the dark for 24 h. The nanoparticles were collected by centrifugation (20 min × 7197 rcf) and re-dispersed in water for three times, in order to remove all the unreacted Rhodamine-B. The purified nanoparticles were then suspended in 2 mL milliQ water and their absorbance was measured via UV-Vis spectroscopy (Fig. S6, ESI†).4.12 Preparation of SPION@DMSA-Mal-PNA
A suspension of 5 mL of SPION@DMSA (0.56 mg mL−1 Fe) was added to a solution of 5 mg of PNA 2 dissolved in 0.5 mL of milliQ water under inert atmosphere. The pH of the reaction mixture was adjusted to 7 and the reaction carried out under ultrasonic irradiation for 4 h at room temperature. The nanoparticles were then collected with a permanent magnet, re-suspended in 5 mL water and centrifuged (3×, 15 min at 7197 rcf). The isolated SPION were then stored under nitrogen atmosphere.Acknowledgements
The authors thank CARIPLO Foundation for having supported this research (project no. 2013-0752, project title “New biomimetic tools for miRNAtargeting”).Notes and references
- (a) J. Huang, Y. Li, A. Orza, Q. Lu, P. Guo, L. Wang, L. Yang and H. Mao, Adv. Funct. Mater., 2016, 26, 3818–3836 CrossRef CAS PubMed; (b) C. Blanco-Andujar, A. Walter, G. Cotin, C. Bordeianu, D. Mertz, D. Felder-Flesch and S. Begin-Colin, Nanomedicine, 2016, 11, 1889–1910 CrossRef CAS PubMed; (c) H. Liu, J. Zhang, X. Chen, X.-S. Du, J.-L. Zhang, G. Liu and W.-G. Zhang, Nanoscale, 2016, 8, 7808–7826 RSC; (d) Y. Piñeiro, Z. Vargas, J. Rivas and M. A. Lõpez-Quintela, Eur. J. Inorg. Chem., 2015, 27, 4495–4509 CrossRef; (e) D. Ling, N. Lee and T. Hyeon, Acc. Chem. Res., 2015, 48, 1276–1285 CrossRef CAS PubMed; (f) L. Zhang, W. Dong and H. Sun, Nanoscale, 2013, 5, 7664–7684 RSC; (g) E. Amstad, M. Textor and E. Reimhult, Nanoscale, 2011, 3, 2819–2843 RSC; (h) A. Figuerola, R. Di Corato, L. Manna and T. Pellegrino, Pharmacol. Res., 2010, 62, 126–143 CrossRef CAS PubMed; (i) M. Mahmoudi, V. Serpooshan and S. Laurent, Nanoscale, 2011, 3, 3007–3026 RSC; (j) A. G. Roca, R. Costo, A. F. Rebolledo, S. Veintemillas-Verdaguer, P. Tartaj, T. González-Carreño, M. P. Morales and C. J. Serna, J. Phys. D: Appl. Phys., 2009, 42, 224002 CrossRef; (k) C. C. Berry, J. Phys. D: Appl. Phys., 2009, 42, 224003 CrossRef; (l) Q. A. Pankhurst, N. T. K. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef; (m) J. Gao, H. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097–1107 CrossRef CAS PubMed; (n) S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- (a) P. Arosio, J. Thévenot, T. Orlando, F. Orsini, M. Corti, M. Mariani, L. Bordonali, C. Innocenti, C. Sangregorio, H. Oliveira, S. Lecommandoux, A. Lascialfari and O. Sandre, J. Mater. Chem. B, 2013, 1, 5317–5328 RSC; (b) V. Amendola, M. Meneghetti, O. M. Bakr, P. Riello, S. Polizzi, D. H. Anjum, S. Fiameni, P. Arosio, T. Orlando, C. D.-J. Fernandez, F. Pineider, C. Sangregorio and A. Lascialfari, Nanoscale, 2013, 5, 5611–5619 RSC; (c) N. Lee, Y. Choi, Y. Lee, M. Park, W. K. Moon, S. H. Choi and T. Hyeon, Nano Lett., 2012, 12, 3127–3131 CrossRef CAS PubMed; (d) E. Poselt, H. Kloust, U. Tromsdorf, M. Janschel, C. Hahn, C. Maßlo and H. Weller, ACS Nano, 2012, 6, 1619–1624 CrossRef CAS PubMed; (e) L. Xiao, J. Li, D. F. Brougham, E. K. Fox, N. Feliu, A. Bushmelev, A. Schmidt, N. Mertens, F. Kiessling, M. Valldor, B. Fadeel and S. Mathur, ACS Nano, 2011, 5, 6315–6324 CrossRef CAS PubMed; (f) M. F. Casula, P. Floris, C. Innocenti, A. Lascialfari, M. Marinone, M. Corti, R. A. Sperling, W. J. Parak and C. Sangregorio, Chem. Mater., 2010, 22, 1739–1748 CrossRef CAS.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- (a) G. F. Goya, V. Grazu and M. R. Ibarra, Curr. Nanosci., 2008, 4, 1–16 CrossRef CAS; (b) K. Maier-Hauff, R. Rothe, R. Scholz, U. Gneveckow, P. Wust, B. Thiesen, A. Feussner, A. von Deimling, N. Waldoefner, R. Felix and A. Jordan, J. Neuro-Oncol., 2007, 81, 53–60 CrossRef CAS PubMed; (c) M. Mallory, E. Gogineni, G. C. Jones, L. Greer and C. B. Simone, Crit. Rev. Oncol. Hematol., 2016, 97, 56–64 CrossRef PubMed.
- (a) A. Bhirde, J. Xie, M. Swierczewska and X. Chen, Nanoscale, 2011, 3, 142–153 RSC; (b) M. Mahmoudi, H. Hosseinkhani, M. Hosseinkhani, S. Boutry, A. Simchi, W. S. Journeay, K. Subramani and S. Laurent, Chem. Rev., 2011, 111, 253–280 CrossRef CAS PubMed.
- (a) Z. P. Xu, Q. H. Zeng, G. Q. Lu and A. B. Yu, Chem. Eng. Sci., 2006, 61, 1027–1040 CrossRef CAS; (b) J. Dobson, Gene Ther., 2006, 13, 283–287 CrossRef CAS PubMed.
- (a) P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS; (b) P. Wittung, P. E. Nielsen, O. Buchardt, M. Egholm and B. Norden, Nature, 1994, 368, 561–563 CrossRef CAS PubMed.
- G. Prencipe, S. Maiorana, P. Verderio, M. Colombo, P. Fermo, E. Caneva, D. Prosperi and E. Licandro, Chem. Commun., 2009, 40, 6017–6019 RSC.
- S. Khadsai, B. Rutnakornpituk, T. Vilaivan, M. Nakkuntod and M. Rutnakornpituk, J. Nanopart. Res., 2016, 18, 263 CrossRef.
- (a) S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed; (b) J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed; (c) F. X. Redl, C. T. Black, G. C. Papaefthymiou, R. L. Sandstrom, M. Yin, H. Zeng, C. B. Murray and S. P. O'Brien, J. Am. Chem. Soc., 2004, 126, 14583–14599 CrossRef CAS PubMed.
- P. Calcagnile, D. Fragouli, I. S. Bayer, G. C. Anyfantis, L. Martiradonna, P. D. Cozzoli, R. Cingolani and A. Athanassiou, ACS Nano, 2012, 6, 5413–5419 CrossRef CAS PubMed.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. B. Na, J. Am. Chem. Soc., 2001, 123, 12798–12801 CrossRef CAS PubMed.
- (a) A. Lassenberger, O. Bixner, T. Gruenewald, H. Lichtenegger, R. Zirbs and E. Reimhult, Langmuir, 2016, 32, 4259–4269 CrossRef CAS PubMed; (b) R. Hufschmid, H. Arami, R. M. Ferguson, M. Gonzales, E. Teeman, L. N. Brush, N. D. Browning and K. M. Krishnan, Nanoscale, 2015, 7, 11142–11154 RSC; (c) K. Woo, J. Hong, S. Choi, H.-W. Lee, J.-P. Ahn, C. S. Kim and S. W. Lee, Chem. Mater., 2004, 16, 2814–2818 CrossRef CAS.
- E. Sevinç, F. S. Ertas, G. Ulusoy, C. Ozen and H. Y. Acar, J. Mater. Chem., 2012, 22, 5137–5144 RSC.
- A. Ruiz, P. C. Morais, R. Bentes de Azevedo, Z. G. M. Lacava, A. Villanueva and M. del Puerto Morales, J. Nanopart. Res., 2014, 16, 2589–2609 CrossRef.
- (a) A. G. Roca, S. Veintemillas-Verdaguer, M. Port, C. Robic, C. J. Serna and M. P. Morales, J. Phys. Chem. B, 2009, 113, 7033–7039 CrossRef CAS PubMed; (b) M. Song, Y. Zhang, S. Hu, L. Song, J. Dong, Z. Chen and N. Gu, Colloids Surf., A, 2012, 408, 114–121 CrossRef CAS.
- O. F. Odio, L. Lartundo-Rojas, P. Santiago-Jacinto, R. Martínez and E. Reguera, J. Phys. Chem. C, 2014, 118, 2776–2791 CAS.
- In TM-AFM, the sample is scanned by an oscillating cantilever whose oscillation amplitude is sensitive to both topography and mechanical properties of the sample surface. A phase angle is associated with the cantilever oscillation. A shift on the phase angle results from differences on AFM tip-sample interactions in which visco-elasticity plays an important role.
- S. Yoshite and Y. Yamada, Eur. J. Biochem., 1979, 101, 395–399 CrossRef.
- G. T. Hermanson, Bioconjugate techniques, 2nd edn, Academic Press – Elsevier, 2008 Search PubMed.
- A. Ruiz, G. Salas, M. Calero, Y. Hernández, A. Villanueva, F. Herranz, S. Veintemillas-Verdaguer, E. Martínez, D. F. Barber and M. P. Morales, Acta Biomater., 2013, 9, 6421–6430 CrossRef CAS PubMed.
- F. Xiong, Z. Y. Zhu, C. Xiong, X. Q. Hua, X. H. Shan, Y. Zhang and N. Gu, Pharm. Res., 2012, 29, 1087–1097 CrossRef CAS PubMed.
- Y. Hou, Y. Liu, Z. Chen, N. Gu and J. Wang, J. Nanobiotechnol., 2010, 8, 25 CrossRef PubMed (14pp).
- L. Tang, T. M. Fan, L. B. Borst and J. Cheng, ACS Nano, 2012, 6, 3954–3966 CrossRef CAS PubMed.
- G. Prencipe, S. Maiorana, P. Verderio, M. Colombo, P. Fermo, E. Caneva, D. Prosperi and E. Licandro, Chem. Commun., 2009, 40, 6017–6019 RSC.
- A. L. Andrade, D. M. Souza, M. C. Pereira, J. D. Fabris and R. Z. Domingues, Cerâmica, 2009, 55, 420–424 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley, 6th edn, 2009 Search PubMed.
- See S. Ghosh and R. Mukhopadhyay, J. Colloid Interface Sci., 2011, 360, 52–60 CrossRef CAS PubMed , and refs therein.
- Details on magnetic hyperthermia theory can be found for example in (a) R. E. Rosenweig, J. Magn. Magn. Mater., 2002, 252, 370–374 CrossRef; (b) L.-M. Lacroix, R. Bel Malaki, J. Carrey, S. Lachaize, G. F. Goya, B. Chaudret and M. Respaud, J. Appl. Phys., 2009, 105, 023911 CrossRef.
- K. Maier-Hauff, F. Ulrich, D. Nestler, H. Niehoff, P. Wust, B. Thiesen, H. Orawa, V. Budach and A. Jordan, J. Neuro-Oncol., 2011, 103, 317–324 CrossRef PubMed.
- A. Lascialfari, M. Filibian, C. Sangregorio and P. Carretta, La Rivista del Nuovo Cimento, 2013, 6, 211–271 Search PubMed.
- (a) R. C. O'Handley, Modern Magnetic Materials Principles and Application, Wiley, New York, 2000 Search PubMed; (b) U. Jeong, X. Teng, Y. Wang, H. Yang and Y. Xia, Adv. Mater., 2007, 19, 33–60 CrossRef CAS.
- L. Lartigue, C. Innocenti, T. Kalaivani, A. Awwad, M. del Mar Sanchez, Y. Guari, J. Larinova, C. Guérin, J. L. G. Montero, V. Barragan-Montero, P. Arosio, A. Lascialfari, D. Gatteschi and C. Sangregorio, J. Am. Chem. Soc., 2011, 133, 10459–10472 CrossRef CAS PubMed.
- L. Kopanja, I. Milosevic, M. Panjan, V. Damnjanovic and M. Tadic, Appl. Surf. Sci., 2016, 362, 380–386 CrossRef CAS.
- The signal intensity SI, and consequently the image contrast in the MRI, is proportional to SI(TR,TE) ρ(1 − e−TR/T1)e−TE/T2, where ρ is the density of tissue nuclear spins, TR the repetition time of the radio frequency (rf) pulse sequence (i.e. the time between consecutive identical sequences) and TE the echo time (i.e. the time between the initial rf pulse and the time at which the signal is received). As a consequence, using the appropriate pulse sequences, it is possible to weight the image on T1 or T2 taking full advantage of the SPION@DMSA property (in detail T1-weighted sequences use short TR and short TE), while T2-weighted sequences use long TR and long TE.
- (a) Y. Gossuin, T. Orlando, M. Basini, D. Henrard, A. Lascialfari, C. Mattea, S. Stapf and Q. L. Vuong, Nanotechnology, 2016, 27, 155706 CrossRef CAS PubMed (11pp); (b) T. Orlando, M. Albino, F. Orsini, C. Innocenti, M. Basini, P. Arosio, C. Sangregorio, M. Corti and A. Lascialfari, J. Appl. Phys., 2016, 119, 134301 CrossRef.
- See for instance: (a) L. He and G. J. Hannon, Nat. Rev. Genet., 2004, 5, 522–531 CrossRef CAS PubMed; (b) J. Krol, I. Loedige and W. Filipowicz, Nat. Rev. Genet., 2010, 11, 597–610 CAS.
- M. V. Iorio and C. M. Croce, EMBO Mol. Med., 2012, 4, 143–159 CrossRef CAS PubMed.
- M. Pittelkow, R. Lewinsky and J. B. Christensen, Org. Synth., 2007, 84, 209–214 CrossRef CAS.
- M. Richter, A. Chakrabarti, I. R. Ruttekolk, B. Wiesner, M. Beyermann, R. Brock and J. Rademann, Chem.–Eur. J., 2012, 18, 16708–16715 CrossRef CAS PubMed.
- O. Ma, M. Lavertu, J. Sun, S. Nguyen, M. D. Buschmann, F. M. Winnik and C. D. Hoemann, Carbohydr. Polym., 2008, 72, 616–624 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00519a |
| This journal is © The Royal Society of Chemistry 2017 |