Phenolate platform for anion exchange in ionic liquids†
Kallidanthiyil Chellappan
Lethesh
,
Dries
Parmentier
,
Wim
Dehaen
and
Koen
Binnemans
*
KU Leuven–University of Leuven, Department of Chemistry, Celestijnenlaan 200F, bus 2404, B-3001, Heverlee, Belgium. E-mail: Koen.Binnemans@chem.kuleuven.be
First published on 27th September 2012
Abstract
Water-soluble ionic liquids can be prepared from halide ionic liquids by a new anion exchange method. This new method for the anion exchange in ionic liquids takes advantage of the strong basicity of phenolate anions (also called phenate anions or phenoxide anions). The principle behind the method is to first prepare the 4-tert-butylphenolate salt of the desired cation, followed by reaction of the 4-tert-butylphenate with a Brønsted acid in a biphasic system formed by water and a water-immiscible organic solvent. The method has been applied to the synthesis of ionic liquids with 1-butyl-3-methylimidazolium, tetrabutylammonium, tetrabutylphosphonium and 1-butyl-1-methylpyrrolidinium cations and a range of anions (formate, acetate, methanesulfonate, tosylate, trifluoroacetate, picolinate, hydrogen dipicolinate, nicotinate, isonicotinate, nitrate, hydrogen sulfate, dihydrogen phosphate). Depending on the nature of the cation, slight modifications of the experimental procedure were required.
Introduction
Ionic liquids (ILs) are solvents that consist of cations and anions.1–4 Typically, they are low-melting organic salts and many of them are liquid at room temperature. Ionic liquids have unique solvent properties, such as an extremely low vapor pressure, an intrinsic ionic conductivity and a wide electrochemical window.5 Their ability to dissolve inorganic and organic compounds, as well as their miscibility with water and organic solvents, strongly depends on the alkyl chain length on the cation and especially on the type of anion. For instance, ionic liquids with the bis(trifluoromethylsulfonyl)imide anion (Tf2N−) are typically immiscible with water and are very useful as an electrolyte for the electrodeposition of reactive metals.6 Ionic liquids with acetate anions, for instance 1-ethyl-3-methylimidazolium acetate, are excellent solvents for cellulose,7 while ionic liquids with sulfonate anions are able to dissolve lignin.8 Most ionic liquids are prepared from precursors with halide counter ions (chloride or bromide), because haloalkanes are typical starting materials for the quaternization reaction.9 The halide counter ions can easily be exchanged via a metathesis reaction by other anions, provided that the starting products are soluble in water and that the resulting ionic liquid separates as a hydrophobic phase from the water. A typical example is the straightforward formation of ionic liquids with the Tf2N− counter ion, which can be synthesized by adding an aqueous solution of LiTf2N to an aqueous solution of a chloride or bromide salt.10,11 The resulting ionic liquid separates from the aqueous phase and lithium chloride remains dissolved in water. Ionic liquids with highly hydrophobic cations can also be prepared by this method.12 In contrast, the synthesis of hydrophilic ionic liquids via a metathesis reaction is considerably more difficult, because the resulting water-miscible ionic liquid will remain in the aqueous phase and no phase separation takes place. The classic method to synthesize hydrophilic ionic liquids was to perform a metathesis reaction using the silver salt of the anion, which results in the precipitation of a silver halide.13 The disadvantages of this method are the high price of silver salts and the risk of contamination of the ionic liquid by silver ions.14 Metathesis reactions with lead salts result in ionic liquids heavily contaminated by lead ions.15 Addition of an alkali metal salt of the anion to a solution of a chloride or bromide ionic liquid in a dry solvent (e.g. dry ethanol or acetone) results in an anion exchange and the precipitation of an alkali salt, but the resulting ionic liquid is often heavily contaminated by halide impurities.16,17 An alternative method for the synthesis is the use of alkylating reagents, e.g. dimethyl sulfate or the methyl ester of triflic acid, to synthesize intrinsically halide-free ionic liquids.18 This method can be extended by transesterification reactions, but the number of anions that can be introduced is limited.19 The alkylating agents can be used as halide-trapping agents, to convert halide ionic liquids into ethyl sulfates, tosylates…20 1-Alkyl-3-methylimidazolium nitrate ionic liquids were prepared by the reaction of 1-methylimidazole with alkyl nitrates,21 and 1,3-dimethylimidazolium dimethyl phosphate was prepared by alkylation of N-methylimidazole with trimethyl phosphate.22 A few reports describe the use of ion exchange resins for the synthesis of hydrophilic ionic liquids.23–25 Unfortunately, this method is hardly suitable for upscaling, due to the relatively low exchange capacity of ion-exchange resins. The anion can be introduced by first transforming the ionic liquid into a hydroxide form, followed by the addition of a Brønsted acid.26–28 However, hydroxide ionic liquids are not easily accessible,29 and the solutions of the hydroxide ionic liquids tend to decompose. Another method is the preparation of an ionic liquid with a methyl carbonate or a hydrogen carbonate anion by using a dialkyl carbonate (and especially dimethyl carbonate) as an alkylating agent, followed by the addition of a Brønsted acid.30–33 The acid decomposes the methyl carbonate or hydrogen carbonate anions and carbon dioxide is released. A disadvantage of the use of dialkyl carbonates as an alkylating agent is their relatively low reactivity, so that reaction temperatures of above 100 °C and high pressures are required.34 Instead of dimethyl carbonate, dimethyl sulfite has been used as the methylating agent.35 The advantage is that much lower reaction temperatures can be used. The main disadvantage is the formation of toxic sulfur dioxide gas. Moreover, dimethyl sulfite is significantly more expensive than dimethyl carbonate. Acetate ionic liquids can be transformed into other ionic liquids by heating these ionic liquids with Brønsted acids or ammonium salts, followed by evaporation of the acetic acid that is formed.36 1-Butyl-3-methylimidazolium ionic liquids were prepared by refluxing 1-butyl-3-methylimidazolium chloride or bromide with an alcohol in the presence of a strong Brønsted acid.37 This method is mainly useful for the preparation of hydrogen sulfate, trifluoromethanesulfonate (triflate) or methanesulfonate ionic liquids.Phenol and its derivatives have been used to extract quaternary ammonium salts from aqueous solutions. Idel et al. describe a process for removing onium salts from aqueous solutions by a adding a phenol to an aqueous solution containing an onium salt, followed by contacting the solution with a water-immiscible organic layer, whereby the onium salt migrates into the organic phase.38 However, this method is a water purification method and has not been extended to anion exchange in organic salts for synthesis purposes. Brannon described how quaternary onium phenolates can be made by the reaction of an aqueous solution of a quaternary onium salt of an inorganic acid (chloride, bromide, iodide) with sodium hydroxide and a phenol.39 Excess phenol is used to act as a solvent for the extraction of the phenolate from the aqueous solution. Separation of the phenol layer from the aqueous layer and removal of the phenol under reduced pressure gives the pure quaternary onium phenolate. The phenolate was further transformed into hydroxide or carbonate salts.
In this paper, we present a new method for the anion exchange in ionic liquids, taking advantage of the strong basicity of phenolate anions (also called phenate anions or phenoxide anions). The principle behind the method is to first prepare the 4-tert-butylphenolate salt of the desired cation, followed by reaction of the 4-tert-butylphenolate with a Brønsted acid in a biphasic system formed by water and a water-immiscible organic solvent. The method has been applied to the synthesis of ionic liquids with 1-butyl-3-methylimidazolium, tetrabutylammonium, tetrabutylphosphonium and 1-butyl-1-methylpyrrolidinium cations and a range of anions. Depending on the nature of the cation, slight modifications of the experimental procedure are required. Because of the important role of phenolate anions in the anion exchange procedure and because of the versatility of the method, we call this method the “phenolate platform”.
Results and discussion
Scheme 1 gives an overview of the optimized process for the preparation of hydrophilic tetrabutylphosphonium and tetrabutylammonium ionic liquids by the phenolate platform. The first step is the synthesis of a hydrophobic ionic liquid precursor, i.e. a phenolate compound, via the metathesis reaction between a halide ionic liquid (with chloride, bromide or iodide anions) and the sodium salt of a phenol derivative. This turned out to be a crucial step, because it determines the amount of halide impurity in the final ionic liquid. The second step is the introduction of the desired anion by the anion-exchange reaction with an aqueous solution of different Brønsted acids to form hydrophilic ionic liquids. 4-Tert-butylphenolate is preferred as the intermediate anion because of its high hydrophobicity, which in turn increases the hydrophobicity of the phenolate intermediate ionic liquid. The hydrophobicity of 4-tert-butylphenolate is very important in the anion-exchange reaction, because the by-product after the reactions with Brønsted acids will be 4-tert-butylphenol, which will stay behind in the organic layer and will not interfere with the newly formed hydrophilic, water-soluble ionic liquids. 4-Tert-butylphenol can easily be removed by decantation/separation of the organic layer (in which the phenol is dissolved) from the water phase.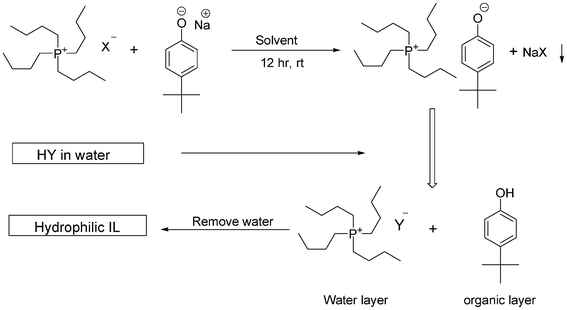 | ||
| Scheme 1 Overall process to prepare hydrophilic ionic liquids by the phenolate method. The tetrabutylphosphonium cation was taken as a model cation. | ||
Initially, the preparation of the phenolate precursors by the extraction of quaternary onium salts from the aqueous phase to a water-immiscible organic solvent by the reaction of the quaternary onion salt with sodium 4-tert-butylphenolate was attempted. Dichloromethane was selected as the organic solvent. In the case of tetrabutylphosphonium and tetrabutylammonium cations, a highly viscous pale yellow liquid was obtained after the extraction and evaporation of the organic layer. However, the 1H NMR spectra revealed that the desired quaternary onium 4-tert-butylphenolate salt was not formed, but rather 4-tert-butylphenol. This may be due to the low polarity of the solvent used for the extraction process. Therefore more polar solvents such as chloroform, 1,2-dichloroethane, ethyl acetate and 2-butanone were used instead of dichloromethane. Ethyl acetate, one of the greener solvents, failed to extract the quaternary onium salts to the organic layer. Just as in the case of extraction with dichloromethane, the 1H NMR spectra showed only the presence of 4-tert-butylphenol in the organic layer. Chloroform, 1,2-dichloroethane and 2-butanone completely extracted tetrabutylammonium and tetrabutylphosphonium salts to the organic layer, to form the corresponding phenolate precursor. Anion exchange reactions with different Brønsted acids were performed and the corresponding ionic liquids were obtained in good yield with low halide content (no precipitate with the AgNO3 test). However, the use of halogenated solvents such as chloroform and 1,2-dichloromethane makes the process less sustainable. Moreover, when the reaction was scaled up (10 g scale), a precipitate was observed with the AgNO3 test. Measurements of the chloride content via the Volhard titration method (vide infra) showed that the chloride concentration in these ionic liquids was as high as 0.4 mol kg−1. The solubility of water in chloroform and 1,2-dichloroethane is about 0.8 wt.%. The solubility of sodium bromide and sodium chloride in water is 36 g/100 mL and 90.5 g/100 mL, respectively. Thus, even a small amount of water in the solvent will drastically change the halide content in the final ionic liquid. Another reason for the higher halide content in the final ionic liquids may be the phase transfer ability of the tetrabutylammonium and tetrabutylphosphonium cations.
To overcome these problems associated with the extraction of the 4-tert-butylphenolate salt from an aqueous to an organic phase, it was decided to prepare the 4-tert-butylphenolate salts in a dry organic solvent. Sodium halide salts have a low solubility in these solvents, so that they precipitate and can be removed from the solution by simple filtration. Dry toluene and dry acetone were employed for the ion exchange reaction of tetrabutylammonium bromide and tetrabutylphosphonium chloride with sodium-4-tert-butylphenolate. In acetone and toluene, both tetrabutylphosphonium phenolate and tetrabutylammonium phenolate were obtained in very good yields (above 90%). Toluene is preferred over acetone because it allows the ion-exchange reaction with aqueous solutions of Brønsted acids to be carried out, without the need for a prior isolation of the corresponding quaternary onium phenolate. Because of the hydrophobicity of 4-tert-butylphenol, it will move to the toluene layer and the hydrophilic ionic liquid formed will migrate to the aqueous layer. Therefore the isolation of the ionic liquid can be achieved by the simple decantation of the toluene layer and the evaporation of the water under reduced pressure. 4-Tert-butylphenol formed after the ion-exchange reaction with acids can be recycled and re-used for the synthesis of new ionic liquid batches. The one-pot procedure with toluene as the solvent for the synthesis of tetrabutylphosphonium and tetrabutylammonium ionic liquids was tested for different Brønsted acids: formic acid, acetic acid, methanesulfonic acid, para-toluenesulfonic acid, trifluoroacetic acid, picolinic acid, dipicolinic acid, nicotinic acid, isonicotinic acid, nitric acid, sulfuric acid, and phosphoric acid. However, in the 1H NMR spectra of the ionic liquids formed, a lower or higher ratio (than the expected value) of the protons of the cations and anions was observed. To obtain a perfect 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between cation and anion, the amount of Brønsted acid added has to be tuned for every specific acid. The amounts of acids used depend on their acidity and polarity. A higher equivalent is needed if the Brønsted acid has a lower polarity and/or a low acidity. It is also worth noting that Brønsted acids with low boiling points (preferably lower than 150 °C, such as acetic acid) can be used in excess for the ion exchange reaction because the free acids can be removed along with water during the isolation of the ionic liquids. The chloride content of the ionic liquids was measured by the Volhard titration method. The 1H NMR spectra showed a complete disappearance of the phenolate anion from the final ionic liquids. Because of the hydrophobic nature of the 4-tert-butylphenol, the side product of the ion-exchange reaction, it does not contaminate the hydrophilic ionic liquids formed. It was possible to recover 4-tert-butylphenol after the ion exchange reaction and it could be reused for the preparation of sodium-4-tert-butylphenolate in the next stage. The ion exchange reaction is preferably performed in a non-polar organic solvent to avoid a reduction in the yield of the final ionic liquids. Ion exchange reactions in polar solvents lead to lower yields, which are caused by the solubility of these ionic liquids in polar solvents, i.e. a major portion of the ionic liquids formed during the ion exchange reaction would stay in the organic phase together with the 4-tert-butylphenol. An overview of the tetrabutylphosphonium and tetrabutylammonium ionic liquids prepared via the phenolate platform is given in Tables 1 and 2.
:1 ratio between cation and anion, the amount of Brønsted acid added has to be tuned for every specific acid. The amounts of acids used depend on their acidity and polarity. A higher equivalent is needed if the Brønsted acid has a lower polarity and/or a low acidity. It is also worth noting that Brønsted acids with low boiling points (preferably lower than 150 °C, such as acetic acid) can be used in excess for the ion exchange reaction because the free acids can be removed along with water during the isolation of the ionic liquids. The chloride content of the ionic liquids was measured by the Volhard titration method. The 1H NMR spectra showed a complete disappearance of the phenolate anion from the final ionic liquids. Because of the hydrophobic nature of the 4-tert-butylphenol, the side product of the ion-exchange reaction, it does not contaminate the hydrophilic ionic liquids formed. It was possible to recover 4-tert-butylphenol after the ion exchange reaction and it could be reused for the preparation of sodium-4-tert-butylphenolate in the next stage. The ion exchange reaction is preferably performed in a non-polar organic solvent to avoid a reduction in the yield of the final ionic liquids. Ion exchange reactions in polar solvents lead to lower yields, which are caused by the solubility of these ionic liquids in polar solvents, i.e. a major portion of the ionic liquids formed during the ion exchange reaction would stay in the organic phase together with the 4-tert-butylphenol. An overview of the tetrabutylphosphonium and tetrabutylammonium ionic liquids prepared via the phenolate platform is given in Tables 1 and 2.
| Compounda | Tm (°C)b |
|---|---|
| a Abbreviations: Tos = tosylate, Isonic = isonicotinate, Nic = nicotinate, Pic = picolinate, DipicH = hydrogen dipicolinate. b <RT: room temperature ionic liquid | |
| [Bu4P][CH3COO] | 57 |
| [Bu4P][CH3SO3] | 65 (lit.: 62.2)42 |
| [Bu4P][Tos] | 57 (lit: 54–59)43 |
| [Bu4P][Isonic] | 215 |
| [Bu4P][Nic] | 186 |
| [Bu4P][Pic] | 59 |
| [Bu4P][DipicH] | 205 |
| [Bu4P][NO3] | 70 (lit: 70–73)43 |
| [Bu4P][HSO4] | 124 (lit: 122)44 |
| [Bu4P][H2PO4] | 149 |
| [Bu4P][HCOO] | 41 |
| Compounda | Tm (°C) |
|---|---|
| a Abbreviations: Tos = tosylate, Isonic = isonicotinate, Nic = nicotinate, Pic = picolinate, DipicH = hydrogen dipicolinate | |
| [Bu4N][CH3COO] | 90 (lit.: 95–98)43 |
| [Bu4N][CH3SO3] | 78 (lit.: 78)45 |
| [Bu4N][Tos] | 75 (lit.: 70–72)43 |
| [Bu4N][Isonic] | 240 |
| [Bu4N][Nic] | 163 |
| [Bu4N][Pic] | 62 |
| [Bu4N][DipicH] | 219 |
| [Bu4N][NO3] | 114 (lit.: 119)46 |
| [Bu4N][HSO4] | 168 (lit.: 169–173)43 |
| [Bu4N][H2PO4] | 154 (lit.: 151–154)43 |
| [Bu4N][HCOO] | 69 |
The method described above had to be adapted for the synthesis of ionic liquids with 1-butyl-3-methylimidazolium and 1-butyl-1-methylpyrrolidinium cations. The extraction of 1-butyl-3-methylimidazolium and 1-butyl-1-methylpyrrolidinium cations from the aqueous phase to the organic phase by the reaction with sodium 4-tert-butylphenolate was not successful, because 1-butyl-1-methylpyrrolidinium 4-tert-butylphenolate and 3-butyl-1-methylimidazolium 4-tert-butylphenolate were not lipophilic enough to be extracted into the organic phase, consisting of dichloromethane, 1,2-dichloroethane, chloroform or ethyl acetate. In order to increase the lipophilicity of the phenolate intermediate, other sodium phenolates such as sodium 2,4-di-tert-butylphenolate and sodium 2,4,6-tri-tert-butylphenolate were employed for the extraction process. In all these experiments, only the presence of the corresponding phenol was observed in the organic layer after extraction. This indicates that even after increasing the number of carbon atoms in the phenol, the lipophilicity of the phenolate precursor was not sufficient to move it to the organic layer. When imidazolium and pyrrolidinium cations with longer alkyl chains (>12 carbon atoms) were used, the corresponding phenolate precursor ionic liquids were formed and extracted to the organic phase. However the ion-exchange reaction with Brønsted acids in toluene led to the formation of emulsions. The emulsion formation was due to the presence of a hydrophobic cation with surfactant properties. Ion-exchange reactions were carried out in other solvents, namely tert-butyl methyl ether, diethyl ether, n-heptane and cyclohexane, to avoid the emulsion formation. Unfortunately, there were no improvements in the results. Even after prolonged centrifugation (1 h at 3600 rpm), the emulsion could not be broken. Due to the failure of the extraction procedure for 1-butyl-3-methylimidazolium and 1-butyl-1-methylpyrrolidinium cations to the organic phase, a one-pot procedure for the synthesis of hydrophilic ionic liquids via the phenolate platform was developed. The metathesis reaction with sodium 4-tert-butylphenolate in toluene was not successful because of the insolubility of imidazolium and pyrrolidinium cations in toluene. 2-Butanone worked as a good solvent for the metathesis and anion exchange reaction of imidazolium cations with Brønsted acids. The solvent must be as dry as possible to minimize the solubility of the alkali salts (NaCl, NaBr, KCl, KBr) in the solvents. Dissolved water will enhance the solubility of these salts in the organic solvents, and as a consequence the halide impurities can find their way into the final ionic liquid. A schematic presentation of the synthetic procedure for the synthesis of 1-butyl-3-methylimidazolium salts via the phenolate platform is shown in Scheme 2. Table 3 gives an overview of the melting points of the 1-butyl-3-methylimidazolium ionic liquids prepared via this method, and Table 4 details the viscosities and densities of those ionic liquids that are liquid at room temperature.
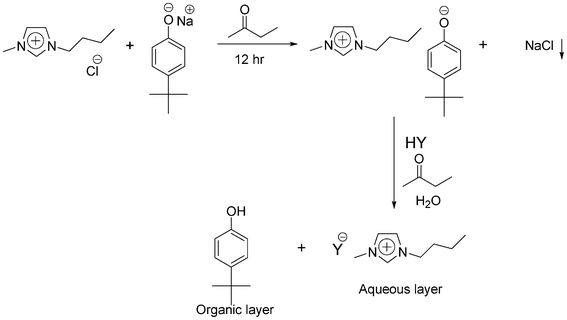 | ||
| Scheme 2 Modification of the phenolate route for the synthesis of hydrophilic ionic liquids with the 1-butyl-1-methylimidazolium cation. | ||
| Compounda | Tm (°C)b |
|---|---|
| a Abbreviations: C4mim = 1-butyl-3-methylimidazolium, Tos = tosylate, Isonic = isonicotinate, Nic = nicotinate, Pic = picolinate. b <RT: room temperature ionic liquid. | |
| [C4mim][CH3COO] | <RT |
| [C4mim][CH3SO3] | 76 (lit.: 74)47 |
| [C4mim][Tos] | 67 (lit.: 67)48 |
| [C4mim][Isonic] | 269 |
| [C4mim][Nic] | 195 |
| [C4mim][Pic] | <RT |
| [C4mim][NO3] | <RT |
| [C4mim][HSO4] | 30 (lit.: 29–32)49 |
| [C4mim][H2PO4] | 165 (lit.: 167)50 |
| [C4mim][HCOO] | <RT |
| [C4mim][CF3COO] | <RT |
| [C4mim][CF3SO3] | <RT |
| Compounda | ρ (g cm−3)b | Viscosity (cP)c | Water content (ppm)d |
|---|---|---|---|
| a C4mim = 1-butyl-3-methylimidazolium, Pic = picolinate. b Density ρ measured at 20 °C. c The viscosities measured at 25 °C. d The water content was measured by coulometric Karl Fischer titration. | |||
| [C4mim][CH3COO] | 1.24 (lit.: 1.243)51 | 150 (lit.: 139.7)51 | 970 |
| [C4mim][Pic] | 1.31 | 1300 | 1100 |
| [C4mim][NO3] | 1.15 lit. 1.15652 | 180 (lit.: 224)52 | 920 |
| [C4mim][CF3COO] | 1.15 (lit. 1.21)10 | 50 (lit.: 73)10 | 900 |
| [C4mim][CF3SO3] | 1.30 (lit.: 1.29)10 | 110 (lit.: 90)10 | 1050 |
The one-pot procedure developed for the 1-butyl-3-methylimidazolium ionic liquids was found to be unsuccessful for the synthesis of 1-butyl-1-methylpyrrolidinium ionic liquids, because of the poor solubility of the 1-butyl-1-methylpyrrolidinium salts in non-polar organic solvents. This insolubility prevented the ion-exchange reaction with Brønsted acids to be carried out without the isolation of 1-butyl-1-methylpyrrolidinium 4-tert-butylphenolate. When the ion-exchange reaction with Brønsted acids were carried out in polar solvents, the yield of the hydrophilic ionic liquids was low, because of their solubility in those solvents. A major part of the ionic liquids stayed in the organic phase. So, a new procedure was developed for the synthesis of hydrophilic ionic liquids based on the 1-butyl-1-methylpyrrolidinium cation via the phenolate route. The metathesis reaction between 1-butyl-1-methylpyrrolidinium bromide and sodium 4-tert-butylphenolate was carried out in dichloromethane. The sodium bromide precipitate was filtered off and dichloromethane was evaporated under vacuum to obtain 1-butyl-1-methylpyrrolidinium 4-tert-butylphenolate in quantitative yield. Both the solvents and starting materials should be as dry as possible in order to minimize the halide contamination in the final ionic liquids. In the second step, ion-exchange reactions with different Brønsted acids were carried out in toluene to introduce the desired anion. A schematic presentation of the synthetic procedure for the synthesis of 1-butyl-1-methylpyrrolidinium salts via the phenolate platform is shown in Scheme 3. Table 5 gives an overview of the melting points of the 1-butyl-1-methylpyrrolidinium ionic liquids prepared via this method. Table 6 lists the densities and viscosities of the ionic liquids that are liquid at room temperature.
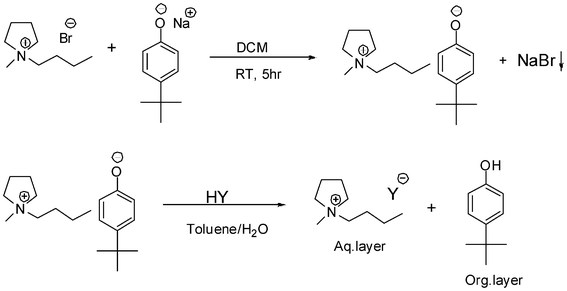 | ||
| Scheme 3 Two-step procedure for the synthesis of hydrophilic ionic liquids with the 1-butyl-1-methylpyrrolidinium cation via the phenolate platform. | ||
| Compounda | Tm (°C)b |
|---|---|
| a Abbreviations: C4mpyr = 1-butyl-1-methylpyrrolidinium, Tos = tosylate, Isonic = isonicotinate, Nic = nicotinate, Pic = picolinate. b <RT: room temperature ionic liquid. | |
| [C4mpyr][CH3COO] | 73 (lit.: 81)53 |
| [C4mpyr][CH3SO3] | 65 (lit.: 63)54 |
| [C4mpyr][Tos] | 114 (lit.: 115)54 |
| [C4mpyr][Isonic] | 227 |
| [C4mpyr][Nic] | 174 |
| [C4mpyr][Pic] | 78 |
| [C4mpyr][NO3] | <RT |
| [C4mpyr][HSO4] | <RT |
| [C4mpyr][H2PO4] | <RT |
| [C4mpyr][HCOO] | <RT |
| [C4mpyr][CF3COO] | <RT |
| [C4mpyr][CF3SO3] | <RT |
| Compounda | ρ (g cm−3)b | Viscosity (cP)c | Water content (ppm)d |
|---|---|---|---|
| a C4mpyr = 1-butyl-1-methylpyrrolidinium, Pic = picolinate. b Density ρ measured at 20 °C. c The viscosities measured at 25 °C. d The water content was measured by coulometric Karl Fischer titration. | |||
| [C4mpyr][NO3] | 1.08 | 160 | 890 |
| [C4mpyr][HSO4] | 1.42 | 800 | 950 |
| [C4mpyr][H2PO4] | 1.33 | 1100 | 1050 |
| [C4mpyr][HCOO] | 1.18 | 90 | 785 |
| [C4mpyr][CF3COO] | 1.10 | 120 | 830 |
| [C4mpyr][CF3SO3] | 1.23 | 180 | 900 |
Extending the phenolate route to other cations such as alkylpyridinium, nitrile-functionalized pyridinium cations and choline chloride was attempted. Unfortunately, none of the procedures described above were successful with those cations. A tarry product was obtained when the different synthetic routes were applied to N-alkylpyridinium and nitrile-functionalized pyridinium cations, because of the decomposition of the pyridine ring. Decomposition of the pyridinium ring has also been observed for trials to prepare pyridinium ionic liquids starting from N-butylpyridinium hydroxide.40 It is can be argued that imidazolium salts are also unstable in strongly basic conditions, resulting in the formation of carbenes. However, these carbenes can be transformed back into imidazolium salts by protonation with a Brønsted acid. In fact, this method can be used to prepare high-purity imidazolium ionic liquids. Extraction of choline chloride from the aqueous phase to the organic phase by the reaction with different sodium phenolates failed, due to the lower lipophilicity of choline chloride. The solubility of choline chloride in organic solvents is very low, so the two-step process also failed to prepare the choline 4-tert-butylphenolate.
It is well known that halide impurities have a very strong influence on the physicochemical properties of ionic liquids.41 Therefore, it is important to determine the halide content in ionic liquids when physicochemical properties have to be reported. All the ionic liquids prepared via the optimized synthetic procedures gave a negative result with the AgNO3 test for halide impurities. The Volhard titration method was used to determine the halide content in the ionic liquid. Seddon et al. reported that the Volhard titration method is a useful tool for the determination of the halide content.41 The Volhard titration method is a wet chemical method for the determination of the halide content. The principle of this method is to precipitate all halide ions present as silver(I) halide by adding a known excess of silver(I) nitrate. The precipitate is collected by filtration, and the excess of silver present in the filtrate is back-titrated with a solution of potassium thiocyanate. This excess of thiocyanate forms a red-brown thiocyanato iron(III) complex with the indicator ammonium iron(III) sulfate. The end point of the titration is determined visually by the first permanent appearance of a red color. The chloride content of the ionic liquids prepared by the optimized synthesis procedures varied between 0.03 and 0.05 mol kg−1. For comparison, tetrabutylammonium acetate was prepared starting from tetrabutylammonium chloride and from tetrabutylammonium bromide. The batch prepared from the chloride salt has a residual chloride concentration of 0.03 mol kg−1, whereas the batch prepared from the bromide salt has a residual bromide concentration of 0.04 mol kg−1. The two ionic liquids thus have a very comparable halide contamination. It is worth noting that the halide content in the ionic liquids prepared by the anion exchange via the phenolate method is much less than that of ionic liquids made by the metathesis reaction of the corresponding sodium salt of the anions, with precipitation of the sodium halide in an organic solvent. For instance, the chloride content in the ionic liquid 1-butyl-3-methylimidazolium nitrate [C4mim][NO3] prepared by the phenolate method is twenty five times lower than the same ionic liquid prepared by conventional metathesis reaction with sodium nitrate (0.04 mol kg−1versus 1.0 mol kg−1).41 In the phenolate method, no additional efforts have to be made to purify the ionic liquid formed after the ion exchange reaction with Brønsted acids. On the other hand, the chloride content in ionic liquid prepared by the metathesis reaction with silver salts is very low (<0.01 mol kg−1), because of the complete metathesis reaction due to the low solubility product of AgCl in aqueous solution. However, silver salts are very expensive. In the phenolate method, the use of expensive chemicals is avoided and low halide contents can be achieved.
Conclusions
In this paper, we described a method for the synthesis of water-miscible ionic liquids, starting from the corresponding halide salts (called “phenolate platform” or “phenolate method”). The new method takes advantage of the strong basicity of phenolate anions. The principle behind the method is to first prepare the 4-tert-butylphenolate salt of the desired cation, followed by the reaction of the 4-tert-butylphenolate with a Brønsted acid in a biphasic system formed by water and a water-immiscible organic solvent. The method has been applied to the synthesis of ionic liquids with 1-butyl-3-methylimidazolium, tetrabutylammonium, tetrabutylphosphonium and 1-butyl-1-methylpyrrolidinium cations with a range of different anions, but the method can be extended to other cations. Depending on the nature of the cation, slight modifications of the experimental procedure were required. 4-Tert-butylphenol could be recycled and it was found that its concentration in the ionic liquids synthesized via this method was below the detecting limit of 1H NMR spectroscopy. However, a necessary condition is that the resulting ionic liquid is highly soluble in water, because the ionic liquid has to be transferred from the organic phase to the aqueous phase. Therefore, this method is not suitable for the synthesis of ionic liquids with highly hydrophobic cations, such as the trihexyltetradecylphosphonium cation, or for ionic liquids with highly hydrophobic anions, such as the bis(trifluoromethylsulfonyl)imide anion. This is not a severe limitation, because these ionic liquids can be prepared by conventional synthesis methods, as they easily separate from the aqueous layer after formation. It was also found that the method cannot be applied to ionic liquids with cations that are not stable against strong bases, such as pyridinium ionic liquids. Imidazolium ionic liquids also have a low stability against strong bases, but they form carbenes that can be transformed back into imidazolium salts by the reaction with a Brønsted acid. Finally, problems were experienced for ionic liquids with choline cations, because of the very low solubility of choline phenolates salts in non-polar organic solvents. The phenolate method allows the preparation of water-miscible (hydrophilic) ionic liquids with a low halide content (0.03 to 0.05 mol kg−1) in comparison with the conventional metathesis reaction with the group I metal salts. The phenolate method is an alternative for the synthesis of ionic liquids by reaction between a methyl carbonate or hydrogen carbonate salt and a Brønsted acid, in cases where the methyl carbonate or hydrogen carbonate salt is not commercially available or not easily accessible via synthetic procedures. From the point of view of green chemistry, our method is of importance, because it provides an easy access to acetate or sulfonate (methanesulfonate, tosylate…) ionic liquids, which are very valuable ionic liquids for the processing of biopolymers, such as cellulose or lignin.Experimental
General information
Elemental analyses (carbon, hydrogen, nitrogen) were conducted on a CE Instruments EA-1110 elemental analyzer. 1H and 13C NMR spectra were recorded on a Bruker Avance 300 spectrometer (operating at 300 MHz for 1H and at 75.5 MHz for 13C). The water content of the ionic liquids was determined by a coulometric Karl Fischer titrator (Mettler Toledo coulometric Karl Fischer titrator, model DL39). The viscosity of the ionic liquids was determined with a Brookfield DV-II + Pro Cone/plate set-up viscometer, with a thermostated sample cell that is being purged with dry nitrogen gas. Differential scanning calorimetry (DSC) measurements were made on a Mettler–Toledo DSC822e module, at a scan rate of 10 °C min−1 in a helium atmosphere. The chloride concentration of the ionic liquids was measured by the Volhard method (see description below). Chemicals were purchased from Acros Organics (Geel, Belgium) or from Sigma-Aldrich (Bornem, Belgium). Extra dry solvents from the AcroSeal® line of Acros Organics were used for experiments with dry solvents. These solvents have a water content of less than 50 ppm. Tetrabutylphosphonium chloride, 1-butyl-3-methylimidazolium chloride and 1-butyl-1-methylpyrrolidinium chloride were purchased from Iolitec (Heilbronn, Germany). All chemicals were used as received, without further purification.Volhard method
An aliquot of the ionic liquid (0.5 g) is accurately weighed, dissolved in deionized water and diluted in a volumetric flask to 250 mL. A solution sample (25 mL) is acidified with nitric acid (6 M, 5 mL) in an Erlenmeyer flask (250 mL) and silver nitrate (0.1 M, 5 mL) is added to precipitate the chloride as silver chloride. After swirling the mixture, the white precipitate was removed by gravity filtration. The precipitate is washed with three portions of dilute nitric acid. To the combined washings, 1 mL of the indicator (a saturated aqueous solution of NH4Fe(SO4)2·12H2O) is added and the solution is titrated with a potassium thiocyanate solution (0.1 M) until the first permanent reddish color occurs. The end point is read from the burette and the procedure is repeated three times. The average of these forms the final result.Synthesis of ionic liquids
The syntheses of tetrabutylphosphonium acetate, 1-butyl-3-methylimidazolium acetate and 1-butyl-3-methylpyrrolidinium acetate are given as typical examples of application of the phenolate platform. Other ionic liquids can be prepared in a similar way by replacing acetic acid by another Brønsted acid. However, the exact amount of acid to be added differs from acid to acid. Therefore, the reader is advised to consult the detailed descriptions of the synthetic procedures that are available as Supplementary Information.†Synthesis of tetrabutylphosphonium acetate
To a solution of tetrabutylphosphonium chloride (10 g, 33.91 mmol) in dry toluene (400 mL) was added sodium 4-tert-butylphenolate (5.83 g, 33.91 mmol). The reaction mixture was stirred vigorously for 12 h and afterwards filtered through Celite. An aqueous solution (500 mL) of acetic acid (2.84 g, 47.47 mmol) was added to the reaction mixture and stirred for 30 min. The organic phase was separated and washed with 50 mL of H2O. The water was removed under vacuum to yield the product as a white solid. Yield: 9.07 g (84%). mp: 57 °C. 1H NMR (300 MHz, D2O): δ = 0.82 (t, 12H), 1.37 (m, 16H), 1.84 (s, 3H), 2.06 (m, 8H). 13C NMR (100 MHz, D2O): δ = 12.55, 17.45 (d), 22.71 (d), 22.88, 23.22 (d), 180.71. CHN elemental analysis for C18H39O2P·2H2O: calculated (%): C 60.98, H: 12.23, found (%): C: 60.63, H: 12.40.Synthesis of 1-butyl-3-methylimidazolium acetate
To a solution of 1-butyl-3-methylimidazolium chloride (10 g, 57.25 mmol) in dry 2-butanone (500 mL) was added sodium 4-tert-butylphenolate (9.85 g, 57.25 mmol). The reaction mixture was stirred vigorously for 12 h and afterwards filtered through Celite. An aqueous solution (500 mL) of acetic acid (5.15 g, 85.87 mmol) was added to the reaction mixture and stirred for half an hour. The organic phase was separated and washed with 50 mL of H2O. The water was removed under vacuum to yield the product as a colorless liquid. Yield: 9.08 g (80%). 1H NMR (300 MHz, D2O): δ = 0.80 (t, 3H; 7.37 Hz), 1.21 (m, 2H), 1.75 (m, 2H), 1.93 (s, 3H), 3.79 (s, 3H), 4.09 (m, 2H), 7.32 (m, 2H), 8.61 (s, 1H). 13C NMR (75 MHz, D2O): δ = 12.59, 18.70, 23.09, 31.21, 35.56, 49.21, 122.17, 123.43, 135.79, 180.75. CHN elemental analysis for C10H18 N2O2·2H2O: calculated (%): C: 51.26, H: 9.46, N: 11.96, found (%): C: 51.20, H: 9.51, N: 12.03.Synthesis of 1-butyl-1-methylpyrrolidinium acetate
To a solution of 1-butyl-1-methylpyrrolidinium 4-tert-butylphenolate (10 g, 34.30 mmol) in toluene (250 mL), an aqueous solution (500 mL) of acetic acid (3.08 g, 51.45 mmol) was added and stirred for 30 min. The organic layer was separated and washed with water (50 mL). The combined aqueous layer was concentrated under vacuum to give 1-butyl-1-methylpyrrolidinium acetate as a colorless solid. Yield (5.66 g, 82%). mp: 78 °C (lit: 81 °C). 1H NMR (300 MHz, D2O): δ = 0.88 (t, 3H, 7.32 Hz), 1.25 (m, 2H), 1.62 (m, 2H), 1.98 (s, 3H), 2.10 (m, 4H), 2.94 (s, 3H), 3.19 (m, 2H), 3.40 (m, 4H). 13C NMR (75 MHz, D2O): δ = 12.86, 19.27, 20.75, 21.33, 25.12, 48.05, 64.11, 64.30, 177.08. CHN elemental analysis for C11H23 NO2·2H2O: calculated (%): C: 55.67, H: 11.47, N: 5.90 found (%): C: 55.59, H: 11.37, N: 5.83.Acknowledgements
This project was supported by the KU Leuven (projects IDO/05/005, GOA 08/05, IOF-KP CO2IL and a DBOF PhD grant to KCL), by the IWT-Flanders (SBO-project “MAPIL”) and by the FWO-Flanders (Research Community “Ionic Liquids”). The authors also acknowledge support by Iolitec (Heilbronn, Germany).References
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef CAS.
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef CAS.
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2011, 13, 2489 RSC.
- Ionic Liquids in Synthesis, ed. T. Welton, P. Wasserscheid, Wiley-VCH, Weinheim, Germany, 2002 Search PubMed.
- P. Bonhote, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Gratzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS.
- P. Nockemann, K. Binnemans, B. Thijs, T. N. Parac-Vogt, K. Merz, A. V. Mudring, P. C. Menon, R. N. Rajesh, G. Cordoyiannis, J. Thoen, J. Leys and C. Glorieux, J. Phys. Chem. B, 2009, 113, 1429 CrossRef CAS.
- J. Kagimoto, S. Taguchi, K. Fukumoto and H. Ohno, J. Mol. Liq., 2010, 153, 133 CrossRef CAS.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc. Chem. Commun., 1992, 965 Search PubMed.
- Ionic Liquids in Synthesis, ed. T. Welton and P. Wasserscheid, 2nd edn, vol. 1, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- J. T. Hamill, C. Hardacre, M. Nieuwenhuyzen, K. R. Seddon, S. A. Thompson and B. Ellis, Chem. Commun., 2000, 1929 RSC.
- J. Fuller and R. T. Carlin, Proc. Electrochem. Soc., 1999, 98, 227 Search PubMed.
- R. E. Del Sesto, C. Corley, A. Robertson and J. S. Wilkes, J. Organomet. Chem., 2005, 690, 2536 CrossRef CAS.
- J. D. Holbrey, W. M. Reichert, R. P. Swatloski, G. A. Broker, W. R. Pitner, K. R. Seddon and R. D. Rogers, Green Chem., 2002, 4, 407 RSC.
- S. Himmler, S. Hormann, R. van Hal, P. S. Schulz and P. Wasserscheid, Green Chem., 2006, 8, 887 RSC.
- P. D. Vu, A. J. Boydston and C. W. Bielawski, Green Chem., 2007, 9, 1158 RSC.
- N. W. Smith, S. P. Gourisankar, J. L. Montchamp and S. V. Dzyuba, New J. Chem., 2011, 35, 909 RSC.
- E. Kuhlmann, S. Himmler, H. Giebelhaus and P. Wasserscheid, Green Chem., 2007, 9, 233 RSC.
- E. Alcalde, I. Dinares, A. Ibanez and N. Mesquida, Chem. Commun., 2011, 47, 3266 RSC.
- E. Alcalde, I. Dinares, A. Ibanez and N. Mesquida, Molecules, 2012, 17, 4007 CrossRef CAS.
- P. Nockemann, B. Thijs, K. Driesen, C. R. Janssen, K. Van Hecke, L. Van Meervelt, S. Kossmann, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2007, 111, 5254 CrossRef CAS.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398 CrossRef CAS.
- Y. Q. Peng, G. Y. Li, J. G. Li and S. J. Yu, Tetrahedron Lett., 2009, 50, 4286 CrossRef CAS.
- J. L. Ferguson, J. D. Holbrey, S. Ng, N. V. Plechkova, K. R. Seddon, A. A. Tomaszowska and D. F. Wassell, Pure Appl. Chem., 2012, 84, 723 CrossRef CAS.
- S. Himmler, A. Konig and P. Wasserscheid, Green Chem., 2007, 9, 935 RSC.
- J. D. Holbrey, R. D. Rogers, S. S. Shukla and C. D. Wilfred, Green Chem., 2010, 12, 407 RSC.
- M. Smiglak, C. C. Hines and R. D. Rogers, Green Chem., 2010, 12, 491 RSC.
- B. Oelkers and J. Sundermeyer, Green Chem., 2011, 13, 608 RSC.
- M. Fabris, V. Lucchini, M. Noe, A. Perosa and M. Selva, Chem.–Eur. J., 2009, 15, 12273 CrossRef CAS.
- R. Kalb, US Patent, 8075803 B2, 2011 Search PubMed.
- L. Szarvas and K. Massonne, US Patent, 2007/0255064 A1, 2007 Search PubMed.
- K. Massonne, M. Siemer, W. Mormann and W. Leng, US Patent, 2010/0217010A1, 2010 Search PubMed.
- R. X. Ren and J. X. Wu, Org. Lett., 2001, 3, 3727 CrossRef CAS.
- K. Idel, D. Freitag and E. Ostlinning, US Patent, 4487698, 1984 Search PubMed.
- J. L. Brannon, US Patent, 2309691, 1941 Search PubMed.
- J. Kagimoto, K. Fukumoto and H. Ohno, Chem. Commun., 2006, 2254 RSC.
- K. R. Seddon, A. Stark and M. J. Torres, Pure Appl. Chem., 2000, 72, 2275 CrossRef CAS.
- U. Domanska and L. M. Casas, J. Phys. Chem. B, 2007, 111, 4109 CrossRef CAS.
- Sigma-Aldrich online catalogue, http://www.sigmaaldrich.com.
- M. De Giorgi, D. Landini, A. Maia and M. Penso, Synth. Commun., 1987, 17, 521 CrossRef CAS.
- H. Nakamoto and M. Watanabe, Chem. Commun., 2007, 2539 RSC.
- M. H. Keshavarz, J. Hazard. Mater., 2006, A138, 448 CrossRef.
- M. Blesic, M. Swadzba-Kwasny, T. Belhocine, H. Q. N. Gunaratne, J. N. C. Lopes, M. F. C. Gomes, A. A. H. Padua, K. R. Seddon and L. P. Rebelo, Phys. Chem. Chem. Phys., 2009, 11, 8939 RSC.
- Y. Liu, L. Liu, Y. Lu and Y. Q. Cai, Monatsh. Chem., 2008, 139, 633 CrossRef CAS.
- P. Wasserscheid, M. Sesing and W. Korth, Green Chem., 2002, 4, 134 RSC.
- M. Yoshizawa-Fujita, K. Fujita, M. Forsyth and D. R. MacFarlane, Electrochem. Commun., 2007, 9, 1202 CrossRef CAS.
- G. McHale, C. Hardacre, R. Ge, N. Doy, R. W. K. Allen, J. M. MacInnes, M. R. Bown and M. I. Newton, Anal. Chem., 2008, 80, 5806–5811 CrossRef CAS.
- B. Mokhtarani, A. Sharifi, H. R. Mortaheb, M. Mirzaei, M. Mafi and F. Sadeghian, J. Chem. Thermodyn., 2009, 41, 1432 CrossRef CAS.
- M. Yoshizawa-Fujita, K. Johansson, P. Newman, D. R. MacFarlane and M. Forsyth, Tetrahedron Lett., 2006, 47, 2755 CrossRef CAS.
- J. Golding, S. Forsyth, D. R. MacFarlane, M. Forsyth and G. B. Deacon, Green Chem., 2002, 4, 223 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra22304j |
| This journal is © The Royal Society of Chemistry 2012 |