Thick mesostructured films via light induced self-assembly†
Héloïse De
Paz-Simon
a,
Abraham
Chemtob
*a,
Florian
Crest
a,
Céline
Croutxé-Barghorn
a,
Laure
Michelin
b,
Loïc
Vidal
b,
Séverinne
Rigolet
b and
Bénédicte
Lebeau
b
aLaboratory of Macromolecular Photochemistry and Engineering, University of Haute-Alsace, ENSCMu, 3, rue Alfred Werner, 68093, Mulhouse Cedex, France. E-mail: abraham.chemtob@uha.fr
bInstitute of Mulhouse Material Science, CNRS, LRC 7228, University of Haute-Alsace, 3 rue Alfred Werner, 68093, Mulhouse Cedex, France
First published on 9th October 2012
Abstract
As an alternative to solvent evaporation, we show that UV light can serve as a stimulus to promote self-assembly during the formation of surfactant-templated silica films. Despite practical and conceptual advantages, the Light Induced Self-Assembly (LISA) route has been poorly described in comparison with its evaporation induced analogue. In this article, we give an exhaustive overview of the potentiality of this photoinduced process using a range of PEO-b-PPO-b-PEO amphiphilic triblock copolymers, and investigating systematically the structural (29Si MAS NMR, XRD) and textural properties (TEM, N2 sorption) of the resultant worm-like mesostructured films. Practically, UV irradiation is applied directly to isotropic micrometric films consisting of a nonhydrolyzed alkoxide-amphiphilic copolymer-photoacid generator mixture, obviating the addition of solvent and water. Photoliberation of Brønsted photoacid generates on demand hydrophilic silica species, driving the micellization of the copolymer. The process is single-step and fast, yielding transparent mesostructured films within a few seconds. A target addressed in this feature is to investigate the film structural evolution resulting from the UV irradiation by in situ Fourier transformed infrared analysis (FTIR). Without the interference of solvent, this powerful technique provides a new perspective on the course of the sol–gel process in mesostructured films. For the first time, hydrolysis is assessed kinetically as well as the silica network condensation and the water content of the film.
Introduction
Mesoporous inorganic and hybrid films have attracted a growing interest due to their potential in high impact applications such as filtration, catalysis, adsorption and optics.1 Their properties generally result from the complex interplay between film thickness, porosity, mesopore size and crystalline phase.2 By its ability to control simultaneously all these physical and structural characteristics, the Evaporation Induced Self-Assembly3–5 (EISA) process has rapidly gained in popularity and earned recognition as the reference methodology of preparing mesostructured films. The first step consists of the preparation of a sol solubilizing a surfactant. The latter isotropic solution is then cast and the resultant evaporation of the volatile components causes two major events forming the cornerstones of the EISA approach: the formation of self-assembled surfactant mesophases and the consolidation of the inorganic network through condensation reactions. Nevertheless, the EISA versatility is plagued by the necessity of using high amounts of organic solvent (usually an alcohol), which remains essential for: (1) the sol preparation, (2) the surfactant solubilization; (3) triggering the micellization upon evaporation; and (4) minimizing the siloxane condensation rate to promote the mesophases ordering. Environmental concern but also the importance of an improved control over mesostructuration prompted us to develop an alternative process in which the self-assembly could be controlled by UV light instead, and eliminating the need of solvent.6 As far as we are aware, there are only two other examples addressing the use of sol–gel photopolymerization for mesoporous silica film synthesis7,8 in a protocol substantially different from the one described here. This relative lack of research is somehow surprising, because a Light Induced Self-Assembly (LISA) process is conceptually and practically simpler than the conventional EISA approach.Our LISA procedure, depicted schematically in Fig. 1, includes non-volatile constituents exclusively and relies on a photoinduced self-assembly catalyzed by photoacid. More specifically, a stable homogeneous solution comprising an alkoxy silica oligomeric precursor, an amphiphilic copolymer and a photoacid generator (PAG) is deposited in the place of a sol, in the form of a film of controlled thickness. Under UV exposition, Brønsted superacids are liberated within the film through PAG photolysis. The low-polymerized hydrophilic silicate species resulting from the photoinduced sol–gel process causes a polarity increase, which eventually drives in situ micellar self-assembly of the copolymer, and ultimately the formation of hybrid mesostructures. In this case, no organic co-solvent was needed, since both the precursor and the amphiphilic copolymer are selected such as to be fully miscible. However, the copolymer is not soluble in the polymerized siloxane network, which is an essential criterion for the surfactant structuration. Water, which is essential for hydrolysis, is not added but provided continuously through atmospheric moisture permeation into the film.9 One indirect advantage of using a non diluted medium is the formation of micrometer thick films whereas mesoporous films of only several hundred nanometers are generally reported in the literature.10 In addition, the catalytic efficiency of the photogenerated acids implies much faster reaction kinetics and well-condensed films with no further need for thermal or chemical post treatment.11
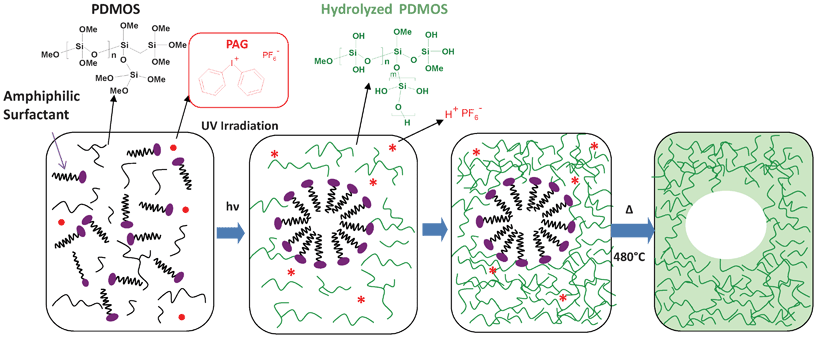 | ||
| Fig. 1 Schematic pathway for the preparation of mesoporous silica films via a light induced self-assembly process. The starting precursor is polydimethoxysiloxane (PDMOS) a fully methoxy silicate oligomer precursor derived from tetramethoxysilane (TMOS). The UV absorption by the iodonium salt (Φ2I+PF6−) affords a set of homolytic and heterolytic cleavages yielding protonic Brønsted superacids of the structure H+PF6. | ||
Herein, a range of silica-block copolymer mesostructured hybrid films have been synthesized by the LISA-derived method. Such a pathway can be implemented with a wide composition of amphiphilic triblock copolymer templates consisting of a relatively hydrophobic poly(propylene oxide) (PPO) middle block and two hydrophilic poly(ethylene oxide) (PEO) end blocks: PEOn-b-PPOm-b-PEOn. Our approach starts with the extensive structural (XRD, 29Si MAS NMR) and textural (TEM, N2 sorption) characterization of the photogenerated mesoporous silica films typically exhibiting a regular worm-like structure. As the process takes place in dry conditions, i.e. without the direct addition of water, special attention was given to the role of water to account for the lack of mesoscopic organization. A second target addressed in this feature is to investigate the film structural evolution resulting from the UV irradiation by in situ Fourier transformed infrared analysis (FTIR). Time-resolved FTIR is a valuable technique, rarely reported, to analyze the fast change in chemical composition of the film during the dual micellization and photoinduced sol–gel process.12–16 Without the interference of solvent prone to saturate the IR signal in EISA, this technique provides a new perspective on the course of the sol–gel process in mesostructured films. For the first time, hydrolysis was assessed kinetically as well as other processes such as the silica network condensation and the water content of the film.
Experimental section
Film preparation
Amphiphilic block copolymers (Pluronic®, HO(CH2CH2O)n(CHCH3CH2O)m(CH2CH2O)nH) were kindly donated by BASF and used as received. In a typical synthesis performed at room temperature, a variable amount of copolymer template, (25 to 75 wt%), was dissolved in polydimethoxysiloxane (PDMOS, ABCR) prior the addition of a PAG (Φ2I+PF6−, 2 wt%, Sigma Aldrich). The homogeneous resultant was found to be stable in the absence of UV light. PDMOS is a non hydrolyzed oligomeric silicate precursor derived from tetramethoxysilane (TMOS); further details regarding its structure have been published elsewhere.9 The homogeneous alkoxide-template-PAG solution was deposited onto a glass substrate using an Elcometer 4340 automatic film applicator equipped with a wire wound bar to form a non volatile liquid film with an initial thickness of 10 μm. The UV insulation was carried out at room temperature under a UV conveyor using a microwave powered mercury vapour lamp (H bulb, Fusion). The spectral output of this electrodeless microwave UV lamp is relatively similar to that of a conventional medium-pressure mercury lamp. The belt speed of the conveyor was set at 10 m min−1 and the lamp intensity at 100%. In these conditions, for each pass, the UV exposure time is 0.23 s and the emitted light dose is 1.46 J cm−2. (UVA [320–390 nm]: 0.45 J cm−2, UVB [280–320 nm]: 0.42 J cm−2, UVC [250–260 nm]: 0.09 J cm−2and UVV [395–445 nm]: 0.50 J cm−2). The samples were subjected to 10 successive passes under the conveyor to yield transparent hybrid solid films. During UV irradiation, the relative humidity (RH) was carefully monitored to be in the range 27–33%. Film calcination was performed without preliminary hydrothermal treatment at 480 °C for 4 h in air.For specific time-resolved FTIR experiments, the formulations were dispensed onto a BaF2 substrate using a bare coater to produce films of similar thickness (10 μm). The in situ IR analysis was performed in transmission configuration (see the characterization section for further details on the spectrophotometric setup), simultaneously with the UV irradiation triggering the polymerization process. In this case, the films were irradiated at a light intensity of 200 mW cm−2 by the polychromatic light of a mercury-xenon lamp (Hamamatsu, L8251, 200 W) fitted with a 365 nm reflector and coupled with a flexible light-guide. The end of the optical guide was placed at a distance of 3 cm from the film and directed at an incident angle of 90° onto the sample window. The sample was maintained in a horizontal configuration with the same composition and experimental conditions as those photopolymerized under the UV conveyor. During this series of experiments, the RH was carefully maintained between 27 and 33%.
Characterization
XRD patterns of the calcined samples were acquired on a Philips X'pert Pro (PANalytical) diffractometer using Cu-Kα radiation (λ = 0.15418 nm; 0.50 < 2θ < 10°; 0.02°/s). TEM micrographs of the calcined films were taken with a Philips CM200 microscope working at 200 kV. Prior to observation, the powdered film was dispersed into water with ultrasound and a few drops of the suspension were deposited at the surface of a copper observation grid. N2 adsorption/desorption isotherms were obtained on a Tristar 3000 (Micromeritics). Calcined samples were first degassed at 150 °C for 4 h. Surface areas (SBET) were determined by the BET method, average pore diameters (DP) were determined from desorption branch by the BdB method.17 Pore wall thicknesses (Wthick) were evaluated from the difference between the d-spacing and pore diameter. To obtain quantitatively reliable 29Si solid state NMR spectra, single pulse magic angle spinning (SPE-MAS) experiments were performed on a Bruker Avance II 300 spectrometer operating at B0 = 7.05 T (Larmor frequency: υ0(Si) = 59.6 MHz) with a Bruker double channel 7 mm probe. The spectrum was recorded using a pulse angle of π/6, a recycling delay of 80 s respectively, a spinning frequency of 4 kHz and high-power proton decoupling during the acquisition. All the NMR experiments were performed at room temperature and chemical shifts reported thereafter are relative to tetramethylsilane. These recording conditions ensured the quantitative determination of the proportions of the different Qn siloxane species. Deconvolution of the spectrum was performed using Dmfit software.18In real-time FTIR experiments, the formulations were simultaneously exposed to UV light and to an IR analytical beam. Such a technique is of interest to assess the sol–gel kinetics throughout the UV irradiation. Infrared spectra obtained in transmission were recorded with a Bruker Vertex 70 spectrophotometer equipped with a liquid-nitrogen-cooled mercury-cadmium telluride (MCT) detector. The spectra were recorded every 0.12 s during the first 18 s then every 5 s from 18 s to 600 s, using a resolution of 4 cm−1. All spectra were baseline corrected prior to integration with the software OPUS 6.5.
Viscosity measurements were performed with a Brookfied DV-II rheometer. Viscosities of pure PDMOS, and hybrid mixture x = 0.25, x = 0.50 and x = 0.75 were respectively of 5, 23, 53 and 96 mPa s.
Results and discussion
I) Photogenerated mesoporous silica film: methodology and characterization
| Pluronic | HLBa | Molecular weight (g mol−1) | PEOn-b-PPOm-b-PEOn | PPO block (%wt) | Soluble in PDMOS | Soluble in water |
|---|---|---|---|---|---|---|
a
 . .
|
||||||
| L81 | 2 | 2750 | PEO3PPO39PEO3 | 90 | Yes | No |
| L121 | 2 | 4400 | PEO5PPO70PEO5 | 90 | Yes | No |
| L62 | 4 | 2500 | PEO6PPO30PEO6 | 80 | Yes | No |
| L43 | 6 | 1850 | PEO6PPO21PEO6 | 70 | Yes | Yes |
| P103 | 6 | 4950 | PEO17PPO60PEO17 | 70 | Yes | Yes |
| P123 | 6 | 5750 | PEO19PPO69PEO19 | 70 | Yes | Yes |
| P84 | 8 | 4200 | PEO19PPO43PEO19 | 60 | Yes | Yes |
| P65 | 10 | 3400 | PEO20PPO30PEO20 | 50 | Yes | Yes |
| P85 | 10 | 4600 | PEO26PPO40PEO26 | 50 | Yes | Yes |
| P105 | 10 | 6500 | PEO37PPO56PEO37 | 50 | Yes | Yes |
| F127 | 14 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
PEO99PPO65PEO99 | 30 | No | Yes |
| F108 | 16 | 14600 |
PEO132PPO50PEO132 | 20 | No | Yes |
 | ||
| Fig. 2 (A) XRD patterns obtained for calcined samples of L121, P123 and P105 at various copolymer/PDMOS wt. ratios: x = 0.25 (dotted line), x = 0.50 (dashed line) and x = 0.75 (plain line). (B) TEM images of the as-calcined films: L121-PDMOS (x = 0.75), P123-PDMOS (x = 0.50), and P105-PDMOS (x = 0.50). | ||
| Template/PDMOS (wt%) | Q2/Q3/Q4a | Condensation degreeb | d spacing (nm) | S BET c (m2 g−1) | V p (cm3 g−1) | D p c (nm) | W thick c (nm) | |
|---|---|---|---|---|---|---|---|---|
a Deconvolution of the spectrum was performed using Dmfit software.18
b
 .
c Surface areas were determined by the BET method, average pore diameters were calculated by the BdB method from desorption branch.17 Pore wall thicknesses were evaluated from the difference between d-spacing and pore diameter. .
c Surface areas were determined by the BET method, average pore diameters were calculated by the BdB method from desorption branch.17 Pore wall thicknesses were evaluated from the difference between d-spacing and pore diameter.
|
||||||||
| L121 | x = 0.25 | — | — | — | 346 | 0.21 | 2.4 | — |
| x = 0.50 | — | — | 6.6 | 500 | 0.38 | 3.1 | 3.5 | |
| x = 0.75 | 1/46/53 | 88% | 7.9 | 489 | 0.46 | 3.8 | 4.1 | |
| P123 | x = 0.25 | 20/57/23 | 76% | — | 269 | 0.03 | 2.2 | — |
| x = 0.50 | 5/67/28 | 81% | 8.2 | 308 | 0.33 | 4.2 | 4.0 | |
| x = 0.75 | 5/51/44 | 85% | 8.2 | 374 | 0.61 | 6.5 | 1.7 | |
| P105 | x = 0.25 | — | — | 8.4 | 219 | 0.14 | 2.6 | 5.8 |
| x = 0.50 | 3/55/42 | 85% | 8.3 | 241 | 0.24 | 4.0 | 4.3 | |
| x = 0.75 | — | — | 8.3 | 356 | 0.51 | 5.7 | 2.6 | |
Nitrogen sorption measurements were also performed, leading to a series of isotherms shown in Fig. 3, obtained for each surfactant at different template/PDMOS ratios. According to IUPAC classification,29 type I isotherm typical of microporous system has been obtained with the P123 surfactant for x = 0.25 ratio whereas others samples exhibited type IV isotherms confirming the mesoporosity. Textural parameters such as the specific surface (SBET), the pore volume (Vp) and the pore diameter (Dp) were extracted from these isotherms and gathered in Table 2. Whatever the surfactant, an increased surfactant concentration generates a higher number of pores which are also larger in size, resulting in thinner walls. For the nonionic amphiphilic copolymer, pore size is not only controlled by the hydrophobic PPO block length (m) but also by its hydrophilic PEO block (n) as only a portion of the PEO chain interacts with the silica network forming the future walls.30–33 A very short PEO block (n = 5) of L121 generates the smallest pores (∼2 nm) as the main contribution for the hydrophobic internal zone results only from the PPO block (m = 70) in this case. As expected, larger pores (4–6.5 nm) were obtained with P123 endowed with a comparable PPO block (m = 69) but a longer PEO block (n = 19). Compared with P105 (m = 56, n = 37), the longer PPO block of P123 is likely to reinforce the hydrophobicity of the PEO chains even if these latter are shorter, accounting presumably for the pore diameter expansion with P123.24
 | ||
Fig. 3 N2 adsorption/desorption isotherms of calcined films obtained with various surfactants (P123, L121, P105) at three surfactant/PDMOS weight ratios: x = 0.25 (■), x = 0.50 ( ) and x = 0.75 ( ) and x = 0.75 ( ). ). | ||
The essence of this reasoning can be captured by the evolution of several textural parameters: first, the fact that the mesostructure only arises at high template concentrations (x ≥ 0.50). Second, the size of the mesopores remains low (<6 nm) compared to other mesoporous systems based on block copolymers41 (∼7–10 nm with P123), showing that a contracted final mesostructure is preferentially built up. Finally, using the intensity of XRD peaks as a probe for order, we clearly identify that P105, having the longest PEO block, improves substantially the nanosegregation without being able to induce an organization. In addition to its thermodynamical effect, Grosso et al.2 suggested an additional role for water: retained in the film, the residual water could confer to the medium fluidity essential to the transition from disordered to ordered micelles. In our water-poor system, the lack of mobility combined with a fast UV-induced gelation is obviously not favourable to the effective conduct of this transition, thus inhibiting the organization of the template in a liquid crystal phase. Consequently, the ability of the block copolymers to exhibit much richer structural polymorphism and to form a great variety of lyotropic phases would require a higher hydration of the silica phase. A high RH is known to promote a higher water concentration inside the film, while a low RH encourages water to evaporate. Further studies using a variety of experimental parameters (RH, light intensity, nature of the surfactant, etc.) are now underway to create the conditions for progressing toward periodically ordered mesostructures.
II) In situ study of sol–gel process in mesostructured hybrid films by real-time FTIR
In situ real-time FTIR experiments were performed to shed light onto the complex and concomitant processes triggered by UV irradiation. There are very few studies reporting the use of this time-resolved technique to study the change in chemical composition of mesostructured silica films.12–16 Practically, in EISA the acquisition of IR spectra starts just after the casting of a droplet consisting of an alkoxide–alcohol–water–HCl mixture. Such a procedure, inherent in this methodology, hinders the study of the sol–gel process in its entirety: first, the handling of a sol implies a hydrolysis already highly advanced or even complete; second, the strongly diluted medium causes signal saturation during several tens of seconds, before the evaporation of the solvent. Consequently, the dynamic studies obtained by RT-FTIR in the literature have not exploited the excellent resolution time (less than 0.2 s) and have essentially focused on a part of the multiple processes occurring: the solvent evaporation kinetics and the qualitative observation of the silica network consolidation. A notable feature that distinguishes our approach is the solvent-free, non-hydrolyzed and photolatent composition. This permits the UV irradiation to control the sol–gel process and indirectly the mesostructuration to be triggered simultaneously with the analyzing IR beam. The RT-FTIR's interest is therefore substantially enhanced since a complete dynamic survey of the sol–gel polymerization becomes accessible: hydrolysis, condensation and water concentration in the film can be assessed simultaneously throughout the irradiation, so as to build up a reliable pathway. Obviously, in situ FTIR is not really informative about mesostructure and porous texture, but rather translates the temporal change, at microscopic level, in chemical composition of PDMOS.1 Besides this, we suspect that the surfactant concentration as well as its self-organization ability are two important factors impacting the sol–gel kinetics. Fig. 4 plots a selection of 3-dimensional FTIR absorption spectra acquired in situ during irradiation of a P123-PDMOS-PAG sample, with wavenumber, irradiation time and absorbance as the x, y and z axis, respectively. Our present discussion emphasizes three distinct regions that will be commented on separately. | ||
| Fig. 4 In situ time-resolved FTIR spectra on a timescale of 600 s of a PDMOS film containing 50 wt% of P123 under UV irradiation in the range of 2700–3650 cm−1 (A) and 800–1600 cm−1 (B). | ||
 | ||
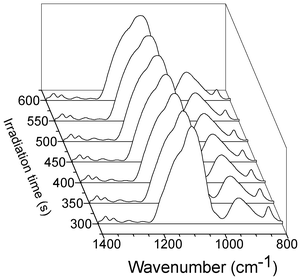
Fig. 5 Methoxy hydrolysis degree during the sol–gel photopolymerization of films containing various ratios (x) in copolymer template: P123 (A) and P105 (B). x = 0 ( , pure PDMOS), x = 0.25 (■), x = 0.50 ( , pure PDMOS), x = 0.25 (■), x = 0.50 ( ) and x = 0.75 ( ) and x = 0.75 (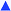 ). ). | ||
 | ||
| Fig. 6 Infrared absorption spectra of the P123-PDMOS film (x = 0.50) in the range 800–1500 cm−1, recorded at different irradiation times after the hydrolysis completion. | ||
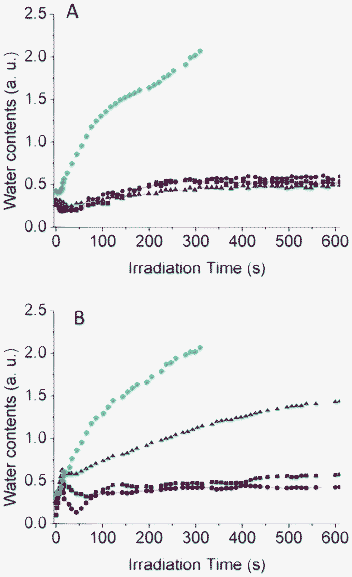 | ||
Fig. 7 Water content evolution during the sol–gel photo-polymerization of P123-PDMOS (A) and P105-PDMOS (B) films containing various ratios in copolymer: x = 0 ( ), x = 0.25 (■), x = 0.50 ( ), x = 0.25 (■), x = 0.50 ( ) and x = 0.75 ( ) and x = 0.75 ( ). ). | ||
Conclusion
We described an alternative and simple approach to silica/surfactant mesostructured films based on a self-assembly process induced by UV light and no longer by solvent evaporation. The light controllable self-assembly relies on the in situ photogeneration of hydrophilic silica species by the release of photoacids, thus driving the partial solubility and micellization of PEOn-b-PPOm-b-PEOn amphiphilic copolymers. The notable features that characterize our photoinduced approach are the speed process, the absence of volatile components (water, organic solvent) and the deposition of a micrometer film consisting of a photolatent oligomeric alkoxide–template–PAG mixture. Our observation emphasized that the absence of water might alter the self-assembly pathway, with a silicate/surfactant mesophase deviating substantially from the theoretical predictions. As recognized for the EISA process, the achievement of organized mesostructures is expected to depend on a number of experimental parameters that will be thoroughly discussed in a subsequent article.Our procedure devoid of solvent, and in which the sol–gel process is fully implemented after deposition, has significantly enhanced the interest of in situ FTIR spectroscopy. RT-FTIR permitted a comprehensive study of the sol–gel process taking place, in particular the hydrolysis kinetics which are normally impeded in a conventional EISA process. We contemplate that in the future this technique should offer interesting opportunities to investigate not only the change in chemical composition but also the general topic of self-assembly of surfactant/silica mesophases.
References
- C. Sanchez, C. Boissière, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682–737 CrossRef CAS.
- D. Grosso, F. Cagnol, G. J. D. A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- H. Yang, N. Coombs, I. Sokolov and G. A. Ozin, Nature, 1996, 381, 589–592 CrossRef CAS.
- H. Yang, A. Kuperman, N. Coombs, S. Mamiche-Afara and G. A. Ozin, Nature, 1996, 379, 703–705 CrossRef CAS.
- H. De Paz, A. Chemtob, C. Croutxé-Barghorn, S. Rigolet and B. Lebeau, Microporous Mesoporous Mater., 2012, 151, 88–92 CrossRef CAS.
- D. A. Doshi, N. K. Huesing, M. C. Lu, H. Y. Fan, Y. F. Lu, K. Simmons-Potter, B. G. Potter, A. J. Hurd and C. J. Brinker, Science, 2000, 290, 107–111 CrossRef CAS.
- S. Nagarajan, J. K. Bosworth, C. Ober, T. P. Russell and J. J. Watkins, Chem. Mater., 2008, 20, 604–606 CrossRef CAS.
- H. De Paz, A. Chemtob, C. Croutxe-Barghorn, S. Le Nouen and S. Rigolet, J. Phys. Chem. B, 2012, 116, 5260–5268 CrossRef CAS.
- G. J. A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093–4138 CrossRef.
- Y. Wan and Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS.
- D. Doshi, A. Gibaud, V. Goletto, L. U. Mengcheng, H. Gerung, B. Ocko, S. Han and C. J. Brinker, J. Am. Chem. Soc., 2003, 125, 11646–11655 CrossRef CAS.
- P. Falcaro, S. Costacurta, G. Mattei, H. Amenitsch, A. Marcelli, M. C. Guidi, M. Piccinini, A. Nucara, L. Malfatti, T. Kidchob and P. Innocenzi, J. Am. Chem. Soc., 2005, 127, 3838–3846 CrossRef CAS.
- P. Innocenzi, P. Falcaro, D. Grosso and F. Babonneau, J. Phys. Chem. B, 2003, 107, 4711–4717 CrossRef CAS.
- P. Innocenzi, T. Kidchob, J. M. Bertolo, M. Piccinini, M. C. Guidi and C. Marcelli, J. Phys. Chem. B, 2006, 110, 10837–10841 CrossRef CAS.
- P. Innocenzi, L. Malfatti, T. Kidchob, P. Falcaro, M. C. Guidi, M. Piccinini and A. Marcelli, Chem. Commun., 2005, 2384–2386 RSC.
- J. C. P. Broekhoff and J. H. de Boer, J. Catal., 1968, 10, 377–390 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- S. Ceccia, E. A. Turcato, P. L. Maffettone and R. Bongiovanni, Prog. Org. Coat., 2008, 63, 110–115 CrossRef CAS.
- M. J. Rosen, Surfactants and Interfacial Phenomena, 3rd edn, 2004 Search PubMed.
- G. Malucelli, R. Bongiovanni, M. Sangermano, S. Ronchetti and A. Priola, Polymer, 2007, 48, 7000–7007 CrossRef CAS.
- S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242 Search PubMed.
- S.-S. Kim, T. R. Pauly and T. J. Pinnavaia, Chem. Commun., 2000, 835–836 RSC.
- E. Prouzet and C. Boissiere, C. R. Chim., 2005, 8, 579–596 CrossRef CAS.
- D. Versace, A. Chemtob, C. Croutxe-Barghorn and S. Rigolet, Macromol. Mater. Eng., 2010, 295, 355–365 CrossRef CAS.
- P. Alexandridis and J. F. Holzwarth, Macromolecules, 1994, 27, 2414–2425 CrossRef CAS.
- P. Petrov, J. Yuan, K. Yoncheva, A. H. E. Muller and C. B. Tsvetanov, J. Phys. Chem. B, 2008, 112, 8879–8883 CrossRef CAS.
- I. Park and T. J. Pinnavaia, Microporous Mesoporous Mater., 2009, 118, 239–244 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- D. Zhao, P. Yang, N. Melosh, J. Feng, B. F. Chmelka and G. D. Stucky, Adv. Mater., 1998, 10, 1380–1385 CrossRef CAS.
- K. Flodström and V. Alfredsson, Microporous Mesoporous Mater., 2003, 59, 167–176 CrossRef.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- P. Kipkemboi, A. Fogden, V. Alfredsson and K. Flodstrom, Langmuir, 2001, 17, 5398–5402 CrossRef CAS.
- B. Lindman and P. Alexandridis, Amphiphilic Block Copolymers Self-Assembly and Applications, Elsevier, Amsterdam, the Netherlands, 2000 Search PubMed.
- M. Sjoberg and T. Warnheim, Surf. Sci. Ser., 1997, 67, 179–205 CAS.
- J. Penfold, E. Staples, I. Tucker and P. J. Cummins, J. Colloid Interface Sci., 1997, 185, 424–431 CrossRef CAS.
- R. Ivanova, B. Lindman and P. Alexandridis, Langmuir, 2000, 16, 3660–3675 CrossRef CAS.
- G. J. A. A. Soller-Illia, E. L. Crepaldi, D. Grosso, D. Durand and C. Sanchez, Chem. Commun., 2002, 2298–2299 RSC.
- E. L. Crepaldi, G. J. A. A. Soler-Illia, D. Grosso, F. Cagnol, F. Ribot and C. Sanchez, J. Am. Chem. Soc., 2003, 125, 9770–9786 CrossRef CAS.
- B. Svensson, U. Olsson and P. Alexandridis, Langmuir, 2000, 16, 6839–6846 CrossRef CAS.
- S. A. El-Safty and J. Evans, J. Mater. Chem., 2002, 12, 117–123 RSC.
- C. Belon, A. Chemtob, C. Croutxé-Barghorn, S. Rigolet, V. Le Houérou and C. Gauthier, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4150–4158 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Figure S1. Effect of the initial film thickness on the XRD patterns of the 75 wt% P123-PDMOS calcined film . See DOI: 10.1039/c2ra21676k |
| This journal is © The Royal Society of Chemistry 2012 |