Palladium catalysed regioselective arylation of indoles, benzofuran and benzothiophene with aryldiazonium salts†
André F. P.
Biajoli
,
Eduardo T.
da Penha
and
Carlos R. D.
Correia
*
Instituto de Quimica, Universidade Estadual de Campinas, Unicamp, Campinas, São Paulo, Brazil, CP 6154 CEP 13084-971. E-mail: roque@iqm.unicamp.br; Fax: 55 19 3521.3023; Tel: 55 19 3521.3086
First published on 1st October 2012
Abstract
Aryldiazonium tetrafluoroborates have been successfully employed in the direct and regioselective arylation of heteroaromatics such as indole, benzofuran and benzothiophene. The cationic aryl palladium intermediates derived from the aryldiazonium salts act as effective electrophiles in the process. These arylating reactions display operational simplicity and provide the arylated heterocycles regioselectively in moderate to excellent yields.
Introduction
Metal catalysed C–C bond forming reactions, such as the cross-coupling reactions, often require the use of pre-functionalized reagents to operate efficiently. These reactions are well established nowadays and have found wide applicability in synthetic organic chemistry.1 In spite of their wide applicability, new protocols bringing greater efficiency and simplicity to these methods are highly desirable. One of those welcome innovations is the so-called “direct C–H arylations”, in which a new Csp2–Csp2 bond is formed between a previously nonfunctionalized olefin or (hetero)arene and an electrophile. A multitude of electrophiles have been employed for C–H arylations.2 Recently, Sanford, König, Matiichuk, and Magedov have used aryl diazonium salts as a source of radicals for the arylation of aromatic and heteroaromatic compounds.3 Besides their intrinsic capability of generating radicals, aryldiazonium tetrafluoroborates are thermostable and easy-to-prepare compounds which have been extensively explored in cross-coupling and Heck reactions.4Non-radical direct C–H arylations of heterocycles using arenediazonium salts are very rare in the literature. To the best of our knowledge, there is only a single report by Colas and Goeldner for the use of aryldiazonium trifluoroacetates in the arylation of benzofuran, as a demonstration of the reactivity of these diazonium salts.5 Therefore, up to this date a systematic study of direct arylation of heteroaromatics with aryldiazonium salts in a non-radical fashion remains undisclosed. In this work, we present examples of highly regioselective “direct arylations” promoted by palladium of indoles, benzofuran, and benzo[b]thiophene employing aryldiazonium tetrafluoroborates. The reactions presented herein proceed under mild conditions and short reaction times. Moreover, reactions were carried out in open flasks, and did not require pre-purification of solvents and/or starting materials.
Results and discussion
Indoles
We initiated our investigation aiming at the arylation of N-methyl indole 1 using 4-methoxyphenyldiazonium tetrafluoroborate 2 in acetic acid or a mixture of acetic acid with other solvents (Table 1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:As shown in Table 1, a maximum yield of 53% was obtained when 1 was treated with 1 equiv. of aryldiazonium salt 2 in the presence of 10 mol% of palladium acetate at room temperature (entry 1). The use of other solvents and catalysts provided the desired product in lower yields. Pd2dba3 performed poorly providing the aryl indole 3 in less than 10% yield. Gratifyingly, the C-2 arylated indole was observed almost exclusively. During these initial investigations we also observed the formation of a diazo product originated from the nucleophilic attack of the aryl indole 3 on the diazonium salt (∼30% yield), which probably explains the moderate yields obtained for aryl indole 3.
We reasoned that if the aryl indole product could be kept out of contact from the remaining aryldiazonium salt, the yields of the desired aryl indole 3 should improve. Treatment of N-methyl indole 1 with one equiv. of aryldiazonium 2 in the biphasic system water/diisopropylether in presence of Pd(OAc)2 at r.t. furnished the desired C-2 aryl indole only in trace amounts (Table 2, entry 1). However, when the same reaction was conducted at 80 °C, a reasonable 68% yield of aryl indole 3 was observed (Table 2, entry 2). Further optimization permitted the synthesis of the C-2 aryl indole 3a in 80–86% yield (entries 7 and 8, Table 2) using 5 or 10 mol% of palladium acetate respectively. A further decrease in the catalyst loading still provided a satisfactory yield of the aryl indole 3a (entry 9). Sterically demanding electron-rich aryldiazonium salts also performed well under the conditions described in Table 2, furnishing the corresponding products 3b and 3c in 72% and 83% yields respectively (entries 10 and 11, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | T/°C | [Pd] (mol%) | t (min) | Yield (%)a |
| a Determined by 1H NMR. b Isolated. c Reaction carried overnight. d 2 equiv. of aryldiazonium salt employed. | |||||
| 1 | 4-OMe | r.t. | 20 | o.n.c | Traces |
| 2 | 4-OMe | 80 | 20 | 20 | 68a |
| 3 | 4-OMe | 50 | 20 | 15 | 54a |
| 4 | 4-OMe | 50d | 20 | 110 | 76a |
| 5 | 4-OMe | 60d | 20 | 55 | 71a |
| 6 | 4-OMe | 40d | 20 | 165 | 77a |
| 7 | 4-OMe | 40d | 10 | 90 | 86 a |
| 8 | 4-OMe | 40d | 5 | 180 | 80 a |
| 9 | 4-OMe | 40d | 2.5 | o.n.c | 68a |
| 10 | 2-OMe | 40d | 10 | 60 | 72b |
| 11 | 2,4-OMe | 40d | 10 | 160 | 83b |
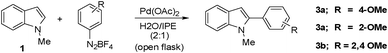
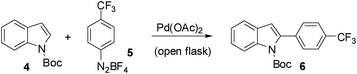
The conditions established in Table 2 were not suitable for electron-deficient aryldiazonium salts due to the extensive formation of diazo products. In order to render the indole system less nucleophilic, we protected the nitrogen atom with the t-butoxycarbonyl (Boc) group. To our surprise, initial attempts to arylate the N-Boc indole 4 with aryldiazonium 5 in the biphasic system water/diisopropylether failed. However, when the arylation was performed in methanol at 65 °C, we obtained the desired aryl indole 6 in 45% yield after 5 min (entry 1, Table 3). At room temperature the yield of 6 decreased to 38% (entry 2). Curiously, when the reactions were carried out in methanol we were also able to isolate the methoxy indole 7 as a side product arising from the addition of methanol to the arylated indole 6 (Fig. 1). The observation of this compound provides some evidence to the mechanism suggested in Scheme 1. To prevent solvent addition we decided to use t-butanol as solvent. In t-butanol aryl indole 6 was obtained in 71% yield after ∼10 h at r.t. (entry 3). Slightly lower yields (65%) were obtained when the reaction was carried out at reflux after only 1 h (entry 4).
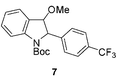 | ||
| Fig. 1 3-Methoxylated 2-arylindole as a side product. | ||
Benzofuran
The results obtained with the indoles encouraged us to extend the direct arylation to other heterocycles like benzofuran. Experiments carried out in the water/diisopropylether system provided the 2-aryl benzofurans 9a and 9b (Table 4) in low yields (<30%). On changing the solvent to methanol we observed somewhat cleaner reactions together with improved yields for the aryl benzofurans 9a and 9b. Therefore, after some experimentation we found out that two distinct protocols worked best for benzofuran arylation depending on the electronic nature of the aryldiazonium salt employed. Electron-rich aryldiazonium salts required reflux of methanol and a four-fold excess of the benzofuran for achieving moderate yields for the C-2 aryl benzofurans (entries 1–3, Table 4). The lower yield (40%) obtained with the 2-methoxyphenyl diazonium salt (entry 3, Table 4) is probably due to some steric effects. On the other hand, arylations with electron-poor and electron-neutral diazonium salts were effective even at r.t., although they required a two-fold excess of the aryldiazonium salts for reasonably good yields of the C2-arylated benzofurans (entries 5 and 7, Table 4). Remarkably, in all cases only the C-2 aryl benzofurans were observed.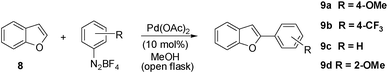
Benzothiophene
For arylations of the benzothiophene, methanol proved to be the solvent of choice: we found out that arylation of benzothiophene could be performed with 4-methoxyphenyl diazonium salt in the presence of Pd(OAc)2 (10–20 mol%) at refluxing methanol to provide aryl benzothiophene 11a regioselectively in an isolated yield of 61% (entry 1, Table 5).|
|
|||||
|---|---|---|---|---|---|
| Entry | R | t (min) | T/°C | [Pd] (mol%) | Yield (%)a |
| a Isolated yields. b 2 equiv. of benzothiophene employed. c 2 equiv. of diazonium salt employed. | |||||
| 1 | 4-OMeb | 50 | 65 | 20 | 61 |
| 2 | 4-OMeb | 180 | 50 | 20 | 74 |
| 3 | 4-OMec | 255 | 50 | 20 | 75 |
| 4 | 4-OMec | o.n. | 50 | 10 | 55 |
| 5 | 4-OMec | 180 | 65 | 10 | 68 |
| 6 | 4-Clc | 60 | 50 | 20 | 85 |
| 7 | 4-Etc | 180 | 50 | 20 | 88 |
| 8 | 2-OMec | 80 | 50 | 20 | 64 |
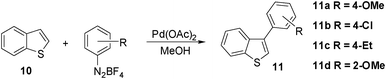
After much experimentation, we found out that a two-fold excess of the aryldiazonium salt, at 50 °C, turned out to be the best conditions for benzothiophene arylation. Gratifyingly, electron-rich (entries 3–5, 8), electron-poor (entry 6) and electron-neutral (entry 7) aryldiazonium salts furnished the corresponding aryl benzothiophenes 11 in good to excellent yields (64% up to 88%). Somewhat surprising, only the C-3 arylated products 11 were observed for the arylation of benzothiophene.
Mechanistic proposal
Concerning a rationale for these arylations, we observed that the reactivity of the substrates seems to correlate with their nucleophilicity.6 This led us to hypothesize that the cationic palladium center might be undergoing nucleophilic attack from the heteroaromatics (Scheme 1), as previously proposed by Zhao.2e Thus, after complexation of the cationic arylpalladium intermediate B to the heterocycle, a few pathways are likely. In the case of benzofuran, a nucleophilic attack on the cationic palladium would generate the benzylic cation C. Proton abstraction restores aromaticity to furnish D, which after reductive elimination generates the aryl benzofurans E and Pd(0). Benzothiophene would react with electrophiles mainly at the C-3 position,7 generating F. Regeneration of aromaticity originates G, which undergoes reductive elimination to produce the C-3 aryl benzothiophenes H. N-Alky indoles would follow a pathway similar to that of benzothiophene to produce the cationic intermediate I. Since only C-2 arylated indoles were observed, an equilibrium involving cations I and J is proposed. Precedents for this type of equilibrium have been previously reported.8 Rearomatization followed by reductive elimination would generate the C-2 aryl indoles M and Pd (0). Another possibility is the rearrangement of I to produce the cationic palladium intermediate K, which ultimately would also lead to the C-2 aryl indoles M and Pd (0).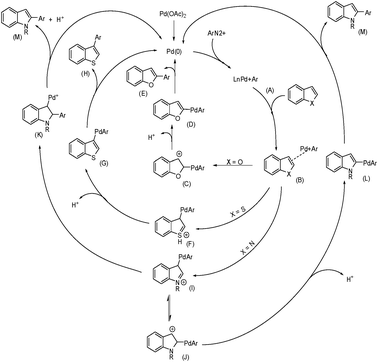 | ||
| Scheme 1 Proposed mechanism for the direct arylation of heterocycles with aryldiazonium salts. | ||
The proposal of an aromatic substitution mechanism finds support in the isolation of the methoxylated product 7. Although unstable, we were able to isolate this compound and assign its structure by NMR experiments (see ESI†). The 2-aryl-3-methoxy indole 7 probably originates from methanol trapping of the intermediate J (Scheme 2). In the specific case of the N-Boc indole arylation using electron-deficient aryldiazonium salts, we hypothesize that the carbonyl of the Boc group might be directing arylation to the C-2 position of the indole. This rationale somehow complements the mechanism proposed in Scheme 1 in the case of the indoles. The capability of carbonyl groups to act as a directing group for cationic aryl palladium species has been pointed out in a recent publication of our group.9
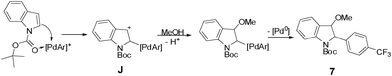 | ||
| Scheme 2 Trapping of intermediate J by methanol to give 2-aryl-3-methoxy indole 7. | ||
Conclusion
In conclusion, we have developed a mild and very simple method for the direct arylation of heterocycles N-methyl indole, N-Boc indole, benzofuran and benzothiophene using aryldiazonium tetrafluoroborates. The method does not involve the generation of radical intermediates and does not require pre-treatment of the substrates or solvent. Moreover, all reactions are carried out in open flasks in the presence of oxygen in wet solvents.10 Electron-rich, electron-deficient, and electron-neutral arenediazonium tetrafluoroborates can be successfully employed leading from moderate to good yields of the arylated heteroaromatic products. Work is ongoing to further extend the scope of these arylations, and to reduce the palladium catalyst loading for some of the reactions.Experimental section
General methods
All reagents were used as received. Column chromatography was performed using a silica gel 200–400 mesh. TLC analysis were performed using silica gel plates. The 1H NMR chemical shifts are reported in δ (ppm) referenced to TMS in chloroform and the residual protons in dichloromethane. 13C NMR chemical shifts were referenced to 13C signals of the solvents. The coupling constants (J) are expressed in Hertz (Hz). IR spectra were recorded using samples that were prepared as either a liquid film between NaCl plates, or pressed in to KBr discs. Melting points are uncorrected. All aryl diazonium salts utilized in this study are known compounds and were prepared according to literature procedures by diazotization of the appropriate aniline.Experimental procedures
Acknowledgements
A.F.P. Biajoli and E. T. da Penha thank CNPq for fellowships.References
- E. Negishi, Handbook of Organopalladium Chemistry, John Wiley & Sons, New York, 2002 Search PubMed.
- (a) C. Hua, S. Dongsheng and Z. Haying, Synlett, 2011, 10, 1472 Search PubMed; (b) A. Strotman, H. Chobanian, Y. Guo, J. He and J. Wilson, Org. Lett., 2010, 12, 3578 CrossRef; (c) N. Deprez, D. Kalyani, A. Krause and M. Sanford, J. Am. Chem. Soc., 2006, 128, 4972 CrossRef CAS; (d) S. Ming, L. Po and K. Yee, Chem.–Eur. J., 2011, 17, 761 CrossRef; (e) J. Zhao, Y. Zhang and K. Cheng, J. Org. Chem., 2008, 73, 7428 CrossRef CAS.
- (a) D. Kalyani, K. McMurtrey, S. Neufeldt and M. Sanford, J. Am. Chem. Soc., 2011, 133, 18566 CrossRef CAS; (b) D. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS; (c) N. Obushak, A. Lesyuk, Y. Gorak and V. Matiichuk, Russ. J. Org. Chem., 2009, 45, 1375 CrossRef CAS; (d) S. Daly, K. Hayden, J. Malik, N. Porch, H. Tang, S. Rogelj, L. V. Frolova, K. Lepthien, A. Kornienko and I. V. Magedov, Bioorg. Med. Chem. Lett., 2011, 21, 4720 CrossRef CAS.
- (a) A. Roglans, P. Anna and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622 CrossRef CAS; (b) J. G. Taylor and C. R. D. Correia, J. Org. Chem., 2011, 76, 857 CrossRef CAS; (c) S. Cacchi, E. Caponetti, M. Casadei, A. Di Giulio and G. Fabrizi, et al. , Green Chem., 2012, 14, 317 RSC; (d) H. Bonin, E. Fouquet and F. X. Felpin, Adv. Synth. Catal., 2011, 353, 3063 CrossRef CAS; (e) J. G. Taylor, A. V. Moro and C. R. D. Correia, Eur. J. Org. Chem., 2011, 8, 1403 CrossRef.
- C. Colas and M. Goeldner, Eur. J. Org. Chem., 1999, 6, 1357 CrossRef.
- H. Mayr and M. Patz, Angew. Chem., Int. Ed. Engl., 1994, 33, 938 CrossRef.
- (a) For examples of regioselective bromination of benzothiophene at C-3, see: L. Yang, Z. Lu and S. Stahl, Chem. Commun., 2009, 6460 RSC; (b) U. Berens, U. Englert, S. Geyser, J. Runsink and A. Salzer, Eur. J. Org. Chem., 2006, 2100 CrossRef CAS; (c) J. F. D. Chabert, L. Joucla, E. David and M. Lemaire, Tetrahedron, 2004, 60, 3221 CrossRef.
- (a) B. S. Lane, M. A. Brown and D. Sames, J. Am. Chem. Soc., 2005, 127, 8050 CrossRef CAS; (b) S. Kirchberg, S. Tani, K. Ueda, J. Yamaguchi, A. Studer and K. Itami, Angew. Chem. Int. Ed., 2011, 50, 2387 CAS.
- P. Prediger, L. Barbosa and C. R. D. Correia, J. Org. Chem., 2011, 76, 7737 CrossRef CAS.
- Arylations were also performed in the absence of palladium to assure the role of palladium as a catalyst. As expected, no arylated products were observed for indoles, benzofuran and benzothiophene in the absence of palladium. Moreover, reactions were also carried out in open reaction flasks or in flasks wrapped up in aluminium foil to block the incidence of light. In all cases, these procedures did not affect the outcome of the reactions. These results, taken together, clearly indicated that a radical mechanism is highly unlikely in these systems.
- L. Wang, W. Yi and C. Cai, Chem. Commun., 2011, 47, 806 RSC.
- S. E. Denmark and J. D. Baird, Org. Lett., 2004, 6, 3649 CrossRef CAS.
- S. Ghosh and J. Das, Tetrahedron Lett., 2011, 52, 1112 CrossRef CAS.
- R. Cano, M. Yus and D. J. Ramón, Tetrahedron, 2012, 68, 1393 CrossRef CAS.
- J. M. Becht, A. Gissot, A. Wagner and C. Mioskowski, Chem.–Eur. J., 2003, 9, 3209 CrossRef CAS.
- N. Takeda, O. Miyata and T. Naito, Eur. J. Org. Chem., 2007, 1491 CrossRef CAS.
- K. Kobayashi, D. Nakai, S. Fukamadi and H. Konishi, Heterocycles, 2010, 81, 1703 CrossRef CAS.
- K. Kobayashi, D. Nakamura and K. Miyamoto, Bull. Chem. Soc. Jpn., 2007, 80, 1780 CrossRef CAS.
- L. Benati, G. Martelli, P. Spagnolo and M. Tiecco, J. Chem. Soc. B, 1969, 4, 472 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra22213b |
| This journal is © The Royal Society of Chemistry 2012 |