Structural trends of ionized water networks: Infrared spectroscopy of watercluster radical cations (H2O)n+ (n = 3–11)†
Kenta
Mizuse
a,
Jer-Lai
Kuo
b and
Asuka
Fujii
*a
aDepartment of Chemistry, Graduate School of Science, Tohoku University, Sendai, 980-8578, Japan. E-mail: asukafujii@m.tohoku.ac.jp; Fax: +81-22-795-6785; Tel: +81-22-795-6572
bInstitute of Atomic and Molecular Sciences, Academia Sinica, Taipei, 10617, Taiwan
First published on 15th February 2011
Abstract
The nature of water networks exposed to ionizing radiation is important in various radiation-related chemistry and biology. To understand structural evolution of ionized water networks at the molecular level, we report here infrared spectra of watercluster radical cations (H2O)n+ (n = 3 − 11) in the gas phase. Spectral features of free OH stretch modes are quite similar to those of protonated waterclustersH+(H2O)n, of which the hydrogen-bond network structures have been revealed. In addition, we observed an extra band attributed to the stretch of an OH radical in (H2O)n+. These results indicate that nominal (H2O)n+ should be regarded as H+(H2O)n−1(OH) motifs having similar network shapes to those of H+(H2O)n. We also analyzed hydrogen-bonded OH stretch bands and found that hydrogen-bond strength is a key factor to determine the position of the OH radical relative to the protonated site (H3O+/H5O2+). Because an OH radical is a weaker hydrogen bond acceptor than water, the first solvation shell of the protonated site is preferentially filled with water. As a result, the OH radical is separated from the protonated (charged) site by at least one water molecule in n ≥ 5 clusters. This result shows the instability of the H3O+-OH ion-radical contact pair in water networks, and implies the higher mobility of the OH radical due to its release from the charged site. Observed structural preferences are confirmed both in cold and warm clusterion sources.
1. Introduction
Interaction between water and ionizing radiation (e.g. X-rays and fast electrons) creates two important radicals, hydroxyl (OH) radicals and hydrated electrons (e−aq).1–3 Because of their high reactivity, ionization of water and subsequent processes play central roles in various radiation-related chemistry and biology in aqueous environments.1–7 One of the most important examples is radiotherapy, in which water in a cancer cell is ionized by radiation and the resulting hydrated electrons and/or OH radicals are thought to cause the DNA strand to break and subsequent cell death.2,3,7Hydrogen(H)-bond network structures including ionization-induced rearrangements are expected to closely relate to chemistry in irradiated water, and then there have been important issues to be solved.2 Despite such importance, not only has the detailed mechanism of ionization processes but also the H-bond network structures around product ions/radicals have remained as open questions.2In the condensed phase, especially in aqueous solutions, chemical reactions following the ionization of water have been extensively studied with various techniques such as ultrafast spectroscopy, and primary processes have been described as eqn (1) and (2):1–3,8–11
| H2O → H2O+ + e− | (1) |
| e− → e−aq | (2a) |
| H2O+ + H2O → H3O+ + OH | (2b) |
For molecular level understanding of structures and dynamics of H-bonded water networks, a hydrated cluster study in the gas phase is a powerful approach.12–86 For example, watercluster anions (H2O)n− have been extensively studied as microscopic models for a hydrated electron in eqn (2a). Photoelectron and optical (infrared (IR) and visible) spectroscopies as well as mass spectrometry have been applied to a broad size range (n ≤ ∼200) of (H2O)n−.15–28,85 In these studies, network structures, electron-binding motifs, and reactivities have been discussed in detail. On the other hand, for the microscopic models of the cationic moiety (eqn (2b)), namely watercluster radical cations (H2O)n+, structural information is still limited as mentioned below. This study focuses on this problem.
Ionization of waterclusters can be regarded as microscopic views of ionization of bulk water and it has been studied with mass spectrometry, photoionization/photoelectron spectroscopy, and theoretical calculations.29–60,86 Although (H2O)n+ is produced in experimental studies, no direct structural information about (H2O)n+ had been reported. Furthermore, it has been pointed out that theoretical calculations often suffer from symmetry-breaking, spin contamination, and/or self-interaction errors in such open-shell doublet systems.57,60 A unique consensus for cluster structures, then, has not been achieved. Even for the simplest case, (H2O)2+, two structural motifs had been suggested.30–32,35–37 One of them is the dimer cation [H2O⋯OH2]+ type and the other is the proton-transferred [H3O+–OH] type. It should be noted that the latter one has the OH radical moiety and corresponds to the exit channel of the reaction in eqn (2b). Very recently, Gardenier et al. have reported IR spectra of argon-tagged (H2O)2+ and have shown that only the proton-transferred [H3O+-OH] type is observed.61 Recent fine theoretical studies, one of which is combined with photoelectron spectroscopy, agree with this result.40,45,46,50–52,57,60,83 Furthermore, Matsuda et al. have identified such proton-transferred type structures in analogous dimer systems such as (NH3)2+ and (CH3OH)2+.62,63 Spectroscopic investigation of (H2O)n+ structures, however, has been limited to the dimer case so far. Though the confirmation of the OH radical creation in the dimer cation is an piece of important progress, the ion-core structure in larger clusters is still ambiguous because the ion-core structure is often changed with solvation. Further experimental studies are also needed to elucidate general trends of network structures and also the interplay between the protonated site and the OH radical under the hydration environment. For larger systems (n ≥ 3), three groups have reported structural motifs from their mass spectrometric analyses, however, these three motifs are different from each other.30–32Fig. 1 shows the structural motifs constructed from their suggestion. In Fig. 1, structure (a) is a solvated “dimer cation core” type, suggested by Yamaguchi et al., and no OH radical exists in this type.32 Both structures (b) and (c) are the proton-transferred type, however, these two differ in the position of the OH radical. In structure (b), suggested by Angel and Stace, the H3O+-OH ion-radical contact pair is fully hydrated via three H-bonds,31 while in structure (c) suggested by Jongma et al., the contact pair dissociates and the OH radical is located at the terminal of the network.30 Structure (a) is expected to show very different reactivity from structures (b) and (c) according to the absence of the OH radical. Furthermore, if the OH radical is formed as in structure (b) and (c), its relative location to the protonated site may be a crucial factor for the reactivity of ionized water networks because the mobility of the OH radical (reactor) may depend on whether the radical is tightly bounded to the protonated (charged) site or not.
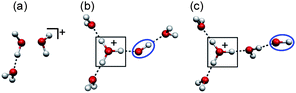 | ||
| Fig. 1 Three different structural motifs of (H2O)n+ (n ≥ 3). These structures were constructed from the previous suggestions.30–32 (See text for details.) The protonated site is indicated by a border. The OH radical moiety is circled. | ||
Structures of (H2O)n+ should reflect the inherent instability of H2O+ and the reactivity of cationized water networks. Such knowledge is quite important to understand ionizing-radiation induced chemistry in water. To reveal structures of (H2O)n+ at the molecular level, we report here IR spectra of (H2O)n+ (n = 3–11) in the OH stretch region. On the basis of the observed spectra, we show structural trends such as network shapes, ion core motifs (“dimer cation” or “proton transferred”), and the existence and location of OH radicals. Relative stability of each possible structure is evaluated by theoretical calculations. Temperature (internal energy) effects are also investigated by changing the condition of the clusterion source and by using the “messenger”65,72 technique.
2. Experimental
IR spectra of (H2O)n+ (n = 3–11) were measured as photodissociation spectra by using a tandem quadrupole mass spectrometer and a coherent IR source.84Watercluster cations were generated in a supersonic jet expansion. The gaseous mixture of H2O (trace) and Ar (5 MPa) was expanded into a vacuum chamber through a high-pressure pulsed valve (Even-Lavie valve).87 The gas pulse was crossed by the electron beam of 200 eV from an electron gun (Omegatron Co.) in the collisional region of the jet. Cluster ions formed were getting larger and were cooled by the following collisions. The resulting cluster ion distribution of this source is shown in Fig. S1† in the supporting information. The cluster ion of interest was selected by the first mass spectrometer (the mass resolution (Δm) was ∼1) and was irradiated by coherent IR light (Laser VisionOPO/OPA). IR spectra were measured by monitoring the photofragment intensity as functions of the IR wavelength. The second mass spectrometer was tuned to select the fragments of both the H2O-loss and the OH-loss channels (Δm ∼ 3). With the higher mass resolution (Δm ∼ 1) measurement, we found that only the OH-loss fragment actually contributes to the observed IR spectra, at least for (H2O)3−5+ (see also the fragment mass spectrum in Fig. S2† in the supporting information). We also measured IR spectra of H+(H2O)n produced in the same ion source for comparison (as shown in the next section). The wavelength of the IR light was calibrated in vacuum wavenumbers (±2 cm−1) by recording an ambient H2O and CO2 vapor spectrum. All the presented IR spectra were normalized by the IR power.For detailed analyses of IR spectra and cluster structures, we carried out density functional theory (DFT) calculations at the MPW1K/6-311++G(3df,2p) level88 using the Gaussian 03 program.89 Calculated structures are visualized by the MOLEKEL program.90 For the (H2O)2+ system, Lee and Kim have reported that this functional gives similar results to the high-level calculations at CCSD(T)/CBS.53 Initial geometries were systematically constructed as mentioned in the following sections. Geometries were fully optimized and harmonic frequency calculations were performed. Calculated frequencies were scaled by a factor of 0.9185. To simulate IR spectra, we used Lorentzian functions with 10 and 50 cm−1 full widths at half maximum for free OH and hydrogen-bonded OH stretch modes, respectively.
Furthermore, we carried out systematic explorations of stable isomers of (H2O)n+ on the B3LYP/6-31+G(d) potential energy surfaces using the GRRM program developed by Ohno and Maeda.80–82,91–93 Details of this method are given in Section 3.7 and in the ESI.†
3. Results and discussion
3.1 Analyses of free OH stretch bands
Fig. 2a shows IR photodissociation spectra of size-selected (H2O)n+ (n = 3 − 11) in the free OH stretch region, and Table 1 collects frequencies of selected bands. In the case of ionic hydrated clusters such as H+(H2O)n, free OH band behavior gives us more structural information on the H-bonded water networks.64–71,73,78 The band patterns in Fig. 2a depend sensitively on the cluster size. It is found that these band patterns of (H2O)n+ are quite similar to those of protonated waterclustersH+(H2O)n (shown in Fig. 2b) reported by several groups so far.64–71,73,78 For H+(H2O)n, the correlation between H-bond network shapes and IR spectra has been well understood (see ref. 94).64–71,73,78,94 Some of the corresponding cluster structures of H+(H2O)n are shown in insets of Fig. 2b. The spectral similarity suggests that (H2O)n+clusters have a similar network shape (oxygen-framework) to that of H+(H2O)n. This structural similarity is in contrast to OH(H2O)n, which is similar to neutral (H2O)n.95 (We note that the structure of H+(H2O)n is quite different from that of neutral (H2O)n at least in the size range considered here.64–71,73,77,78)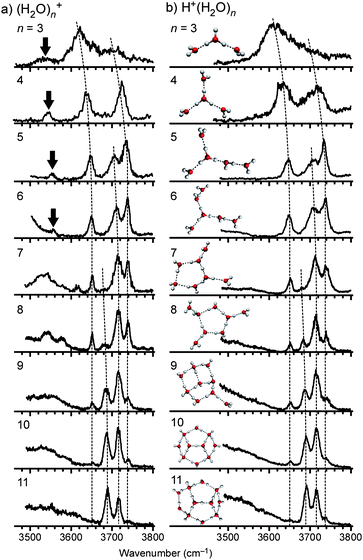 | ||
| Fig. 2 (a) IR spectra of (H2O)n+ (n = 3–11) in the free OH stretch region. (b) IR spectra and representative structures of H+(H2O)n. Dotted curves show each series of bands as assigned in ref. 94. The bands indicated by arrows are the free stretch of the OH radical moiety. | ||
| Freq. (cm−1) | Assignment | Freq. (cm−1) | Assignment |
|---|---|---|---|
| a “HB to ·OH” means H-bonded OH stretch of water towards an OH radical. All the other bands shown here are attributed to free OH stretches of assigned moieties. νsym and νantisym are the symmetric and antisymmetric stretches, respectively. Band frequencies were determined by fitting each band with a single Lorentzian function. Errors in the fitting are shown in parentheses. Absolute accuracy is ±2 cm−1 due to the band width of our IR source (< ∼3 cm−1) and the calibration method. | |||
| (H2O)3+ | (H2O)5+ | ||
| 3536(4) | OH radical | 3458(1) | HB to ·OH |
| 3624(1) | 1-coord (νsym)/H3O+ | 3553(1) | OH radical |
| 3697(3) | 1-coord (νantisym) | 3647(1) | 1-coord (νsym) |
| 3707(1) | 2-coord H2O | ||
| 3734(1) | 1-coord (νantisym) | ||
| (H2O)4+ | (H2O)6+ | ||
| 3103(3) | HB to ·OH | 3496(1) | HB to ·OH |
| 3543(1) | OH radical | 3557(1) | OH radical |
| 3639(1) | 1-coord (νsym) | 3649(1) | 1-coord (νsym) |
| 3725(1) | 1-coord (νantisym) | 3711(1) | 2-coord H2O |
| 3737(1) | 1-coord (νantisym) | ||
The spectra of (H2O)n+ are not identical to those of H+(H2O)n. In the 3500–3600 cm−1 region, an additional band is observed for (H2O)3 − 6+ and it is indicated by the arrow in Fig. 2a. The frequency of this band is too low to be attributed to any free OH stretch of the H2O moieties. Because the gas-phase frequency of the stretch vibration of OH radical is 3570 cm−1,96 we assign this band to the free stretch of an OH radical moiety in the clusters. This assignment is consistent with the OH radical stretch frequencies in (H2O)2+·Ar1,2, 3511 and 3499 cm−1, respectively.61 The presence of the OH radical stretch band as well as the spectral similarity to H+(H2O)n indicates that nominal watercluster cations (H2O)n+ (n ≥ 3) should be regarded as “protonated waterclusters with an OH radical”, H+(H2O)n−1(OH), as has been reported for (H2O)2+.61 In this study, we use the notation, (H2O)n+, for simplicity. These considerations are also a negative evidence for the “dimer ion core” type as in Fig. 1(a). The absence of the “dimer cation core” type is confirmed in Section 3.7 and the supporting information.† For H+(H2O)n−1(OH) type structures, mutual positions of the protonated core and the OH radical, that is, distinction between the two structures in Fig. 1(b) and (c), are essential to characterize cluster structures, implying the reactivity (mobility) of the produced OH radical. In the following, we discuss more detailed structures in each cluster size on the basis of the spectra including the H-bonded OH stretch region.
3.2 (H2O)3+
Fig. 3a shows the IR spectrum of (H2O)3+ in the 2100–4000 cm−1 region. For comparison, the structure and IR spectrum of H+(H2O)3 are shown in Fig. 3c. As was described in the former section, these two spectra are similar to each other, however, the spectrum of (H2O)3+ has additional bands at around 2800 and 3536 cm−1. The latter band, displayed in blue, is attributed to the OH radical stretch. The free OH bands suggest that the structure of (H2O)3+ should resemble that of H+(H2O)3 and have an OH radical moiety. From these requirements, the structural motif of (H2O)3+ is identified as shown in Fig. 3b. The structure in Fig. 3b is constructed by substituting one of the water molecules in H+(H2O)3 with an OH radical. On the basis of this structure, we carried out DFT calculations at the MPW1K/6-311++G(3df,2p) level to obtain the energy optimized structure and its simulated IR spectrum. The calculated IR spectrum shown in Fig. 3b reproduces the band pattern in the free OH stretch region and the band at around 2800 cm−1. This agreement supports the structure shown in Fig. 3b. The calculation indicates that the observed band at around 2800 cm−1 is assigned to the OH stretch of the H3O+ core, hydrogen-bonded to the OH radical moiety. This is reasonable because no clear corresponding peak is observed for H+(H2O)3, which does not contain an OH radical (a broad absorption in the H+(H2O)3spectrum (< ∼3400 cm−1) would be attributed to a long tail of the lower frequency H3O+ stretch band, which has been observed at ∼1880 cm−1 in the Ar-mediated spectrum. (Ref. 72) The drop of this band below 2400 cm−1 can be accounted for by the low dissociation yield due to the lower IRphoton energy). An additional band is seen at around 3150 cm−1 in the (H2O)3+spectrum, and it is absent in the calculated spectrum. We tentatively assigned this band to the overtone of the H3O+ stretch.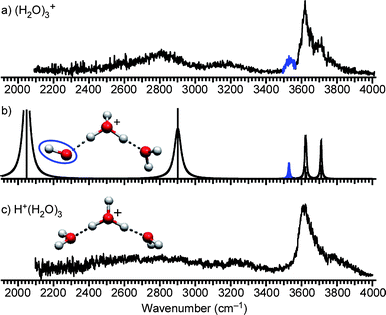 | ||
| Fig. 3 (a) IR spectrum of (H2O)3+. The band highlighted in blue is the free stretch of the OH radical moiety. (b) Calculated cluster structure and corresponding IR spectrum at the MPW1K/6-311++G(3df,2p) level. The OH radical moiety is circled. (c) IR spectrum and structure of H+(H2O)3. | ||
3.3 (H2O)4+
The same analysis as in (H2O)3+ is applicable to (H2O)4+. Fig. 4a shows the experimental IR spectrum of (H2O)4+ and Fig. 4c shows the structure and the spectrum of H+(H2O)4. H+(H2O)4 has the symmetrically solvated H3O+ core (Eigen cation).64,65,72,79 One of the solvating water molecules is replaced with a OH radical, and the plausible (H2O)4+ structure (Fig. 4b) is obtained. By using DFT, we calculated the IR spectrum (Fig. 4b). The simulated IR spectrum is in good agreement with the observed spectrum, verifying the structure in Fig. 4b. The minor extra band at around 3800 cm−1 in the observed spectrum is assigned to a combination band of a free OH stretch and a low-frequency mode as in the previously reported IR spectroscopy of various hydrated clusters including H+(H2O)4 (Fig. 4c).64,67,79 A weak shoulder at around 3380 cm−1 is not expected by the calculation. The tentative assignment is an overtone/combination band or minor contribution from other isomers. The latter would be less plausible because the presence of other isomers affects band patterns in the free OH stretch region.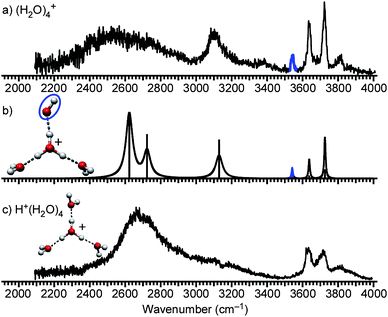 | ||
| Fig. 4 (a) IR spectrum of (H2O)4+. The band highlighted in blue is the free stretch of the OH radical moiety. (b) Calculated cluster structure and corresponding IR spectrum at the MPW1K/6-311++G(3df,2p) level. The OH radical moiety is circled. (c) IR spectrum and structure of H+(H2O)4. | ||
The IR spectrum of (H2O)4+ also provides insight into hydrogen bond strength, which should be an important factor for further structure determination. In the observed spectrum, two H-bonded OH bands are seen. The lower frequency band below 2800 cm−1 corresponds to the similar band in H+(H2O)4, and it is assigned to the H-boded OH stretches toward the water molecules. On the other hand, the higher frequency band at around 3100 cm−1 is characteristic in (H2O)4+ and then it is attributed to the H-bonded OH stretch toward the OH radical. Because a stronger hydrogen bond generally results in a lower OH stretch frequency,72,97,98 the higher frequency of the band at 3100 cm−1 shows that the OH radical is a weaker hydrogen bond acceptor than water. This is consistent with the smaller proton affinity of ·OH (593 kJ mol−1) than H2O (691 kJ mol−1).99 In the following, we show this different hydrogen bond strength is a key to determine the structures of larger-sized clusters. It should be noted that a similar discussion can be applied to (H2O)3+, however, the band of the H-bonded OH toward a water is expected to be located outside of the measurement range (∼2050 cm−1, in Fig. 3b).
3.4 (H2O)5+
Fig. 5a shows the experimental IR spectrum of (H2O)5+ and Fig. 5e shows the structure and the spectrum of H+(H2O)5. Here, two bands highlighted in blue (3553 cm−1) and in red (3458 cm−1) are characteristic in (H2O)5+ and absent in H+(H2O)5, implying they are related to the OH radical moiety. The band in blue is the stretch of the OH radical as in (H2O)3,4+. The band in red (3458 cm−1) is absent in both H+(H2O)n and (H2O)3,4+. The band width is broader than the free OH bands, indicating this is a H-bonded OH stretch band. In addition, its band frequency is the highest among the H-bonded OH bands. This means the band in red is a signature of a weakly H-bonded site.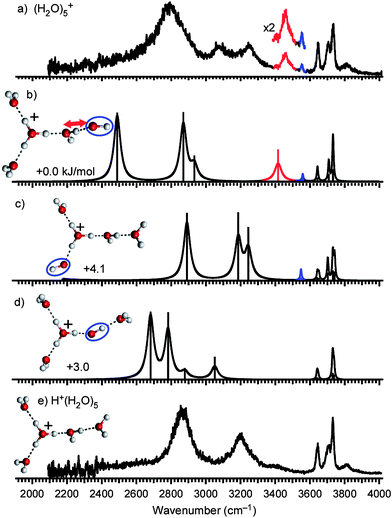 | ||
| Fig. 5 (a) IR spectrum of (H2O)5+. The band highlighted in blue is the free stretch of the OH radical moiety. The red band is the marker of a H-bond from H2O to ·OH (see text). (b–d) Calculated cluster structures and corresponding IR spectra at the MPW1K/6-311++G(3df,2p) level. The OH radical moiety is circled. (e) IR spectrum and structure of H+(H2O)5. | ||
For the structural consideration, the same argument as in (H2O)3,4+ can be applied; however, there is complexity in (H2O)5+. Although only one type of structure has been identified experimentally for H+(H2O)5 (Fig. 5e),67,72,79 three possibilities arise for the corresponding (H2O)5+ (Fig. 5b–d). Structures in Fig. 5b–d differ in the position of the OH radical. In structure I (Fig. 5b), the H3O+–OH ion-radical contact pair is dissociated by a water molecule, and then the OH radical locates at the terminal (surface) of the H-bond network. In structure II (Fig. 5c), the OH radical is also at the terminal, but is directly bound to the H3O+ core. In structure III (Fig. 5d), the H3O+–OH ion-radical contact pair is fully hydrated and the OH radical is inside the network. Here, Structure I and III are the same as those in Fig. 1b and 1c, respectively.
In the following, we examine the plausible spectral carriers and band assignments on the basis of simulated spectra shown in Fig. 5b–d. The contribution of the most stable structure I is obvious because of the appearance of the characteristic red band, which can be seen only in structure I. According to the normal mode analysis, the characteristic red band (3458 cm−1) is attributed to the H-bonded OH stretch of a water molecule toward the OH radical (denoted by the red arrow in Fig. 5). Here this water molecule bridges the H3O+ core and the OH radical. This assignment is reasonable to the weaker H-bond accepting ability of the OH radical, as discussed for (H2O)4+. The calculated spectrum of structure I well reproduces the observed spectrum in the 3500–3800 cm−1 region, but the calculated 2500 cm−1 band is missing in the observed spectrum. This missing band is attributed to the OH stretch of H3O+ toward the 2-coordinated water. The proton toward the 2-coordinated water is partially delocalized to the neighboring water because of the cooperative H-bond to the second solvation shell molecule (the OH radical), and it would have the character of the shared proton. Its transition frequency is, then, expected to be much lower than the present harmonic frequency due to the large anharmonicity. The same argument has been reported for H+(H2O)5.72,79 The calculated spectrum of structure I also does not account for the observed features in the 3000–3200 cm−1 region, and it seems that the significant coexistence of structure II explains these bands. In this case, structure III may also coexist because of its similar relative energy, and its spectrum well overlaps with the observed one. An alternative assignment of the bands are overtones and/or combination bands of structure I. The unexpected band at 3270 cm−1 in the spectrum of (H2O)2+·Ar has been explained by the overtone of the bonded H3O+ stretch.61 Here, we should note that the major contribution of structures II and III causes a serious conflict with the high frequency region of the observed spectrum. The intensity of the red marker band relative to the free OH bands can be reproduced only by the major contribution of structure I. Moreover, we observed that only the loss of the OH radical occurs upon the IR vibrational excitation of the free OH stretch band at 3734 cm−1, which is the vibration of a terminal water and is commonly expected for all the three isomers. The H2O-loss is quite negligible in this excitation (see also Fig. S2†). This suggests that the OH radical lies at the terminal of the network. A hydrogen bond to the OH radical moiety should be the weakest one in the cluster. Because the hydrogen bond with the ion-core is much stronger than those between neutral moieties, the OH moiety should be bound to a neutral moiety, as in structure I. If there is much contribution of structures II and III, the fragmentation pattern should be largely different. These results support that the features at 3000–3200 cm−1 are assigned to overtone/combination bands and therefore structure I is dominant in (H2O)5+. The coexistence of structures II and III cannot be excluded, but they should be minor.
For further confirmation of the dominance of structure I, we examine the energetics. Because clusters formed in the present ion source have a finite temperature (∼180 K, see the section 3.8), energetics including entropy is worth mentioning. We calculated relative Gibbs free energies at the temperature range of 0–400 K (detailed results are given in Fig. S3†). We found that both structures II and III are less stable than structure I at this temperature range. While the relative instability of structure III is constant, that of structure II increases with rising temperature (this would be due to the presence of the low frequency mode in structure I (∼8 cm−1) corresponding to the rearrangement motion of the weakly bounded OH radical. In structure II, the OH radical is tightly bounded to H3O+, and no low frequency modes below 10 cm−1 are found). At 180 K, the relative populations (canonical probabilities) of structure I, II, and III are calculated to be 0.90, 0.04, and 0.06, respectively, under the harmonic vibrational approximation. This evaluation also shows the minor contribution of these isomers.
In structure I, the red marker band is due to the H-bonded OH to the OH radical moiety, and its high frequency reflects the weak ability of the OH radical as a H-bond acceptor. From the viewpoint of the H-bond strength, structure I is rationalized because the first solvation shell of the H3O+ is filled with three water molecules, which can form a stronger H-bond than the OH radical, and the OH radical, weaker H-bond acceptor, is pushed out to the second solvation shell.
Such an ion-radical contact pair separation has also been indicated by the recent ab initio molecular dynamics calculations. Tachikawa has calculated vertical ionization dynamics of (H2O)n≤5 and reported that the second proton transfer occurs after the H3O+–OH pair formation resulting in the separation of the contact pair.41 Similar results have been reported by Novakovskaya46,47 and by Livshits et al.83 Though our clusterion source does not utilize vertical ionization of neutral (H2O)n and detailed processes in the cluster formation are unknown, the reported calculations are consistent with the present results in terms of the separation of the H3O+–OH contact pair.
According to the above discussion, we have shown that the band highlighted in red is due to the “bridging” water weakly H-bonding to the OH radical, and this band is an important signature for the separation between the protonated ion core and the OH radical. On the basis of this knowledge, we analyzed structures of larger clusters.
3.5 (H2O)n+ (n ≥ 6)
To investigate the generality of the separation between the protonated site and the OH radical in larger H-bonded water networks, we analyzed the IR spectra of (H2O)n≥5+ shown in Fig. 6. As clearly seen in the 3500–3570 cm−1 region, the marker band attributed to the “bridging” water moiety remains remarkable in these clusters. This result indicates that the separation between the protonated site and the OH radical is a general trend in larger-sized (H2O)n+.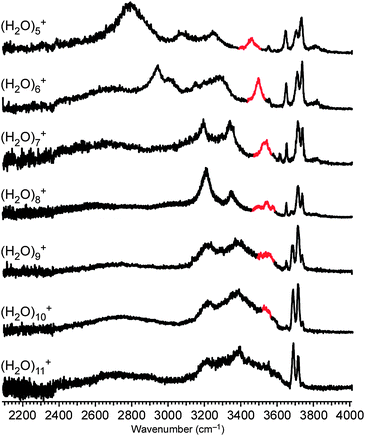 | ||
| Fig. 6 IR spectra of (H2O)5−11+ in the 2100–4000 cm−1 region. The red band is the marker of a H-bond from H2O to ·OH. | ||
It should be noted that spectra of larger-sized (H2O)n+ become more similar to those of H+(H2O)n due to the decrease of the relative weight of the OH radical and the “bridging” water site. As a result, we could not find remarkable differences between the spectra of (H2O)11+ and H+(H2O)11. However, the clear appearance of the marker band and the smooth spectral changes in n ≤ 10 strongly suggest that the trends remain in clusters larger than n = 10.
3.6 Structures of (H2O)n+
We obtained three general trends in structures of (H2O)n+ (n ≥ 3). 1) Nominal watercluster cations (H2O)n+ form proton-transferred type H+(H2O)n−1(OH). 2) Structures (oxygen-frameworks) of the network are quite similar to those of H+(H2O)n. 3) After the first solvation shell of the protonated ion core is completed, the OH radical is separated from the ion core by at least one water molecule. On the basis of these insights, we can easily infer the preferential structures of larger-sized (H2O)n+. Fig. 7 shows the plausible structures of (H2O)6,7+. These structures were constructed from the well known structures of H+(H2O)n67,72,82 by substituting one of the water molecules away from the protonated site with an OH radical. For n ≥ 8, many more isomers are expected even for H+(H2O)n,67,76,81 however, we can deduce their structures by the same procedure.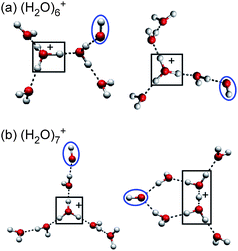 | ||
| Fig. 7 The most plausible structures of (H2O)6,7+, constructed on the basis of experimental findings. The protonated site is surrounded by a border. The OH radical moiety is circled. | ||
These results seem to be very simple but may have an important implication for the ionizing radiation-induced processes in bulk water. As seen in (H2O)n+, the ionization of a water molecule should result in a H3O+–OH contact pair formation via a fast proton transfer. This study demonstrated that this H3O+–OH contact pair is not stable in water networks and the separation between the ion core and the OH radical occurs. In radical reactions, a reaction barrier height is generally quite low and a reaction rate should be determined by the mobility of radicals. It is expected that the OH radical separation from the protonated site would lead to the higher reactivity compared with the case of the contact pair, in which the OH radical is tightly bound to the charged site.
3.7 Other stable structures
We experimentally revealed structures of (H2O)n+; however, in the present study, we did not observe some structures suggested so far, such as dimer cation core type shown in Fig. 1a. It is also valuable to characterize both the observed and other stable structures on the potential energy surface. For this purpose, we carried out systematic explorations of stable isomers of (H2O)3−5+ on the B3LYP/6-31+G(d) potential energy surfaces by using the GRRM program.80–82,91–93 This approach has been applied to H+(H2O)5−8 and has successfully found many local minima including the experimentally identified ones.81,82 We used this program with similar parameters to those reported for H+(H2O)n (for more details, see the ESI†).81,82 We obtained 3, 15, and 75 stable isomers for (H2O)3+, (H2O)4+, and (H2O)5+, respectively. After the reoptimization at the MPW1K/6-311++G(3df,2p) level, we found that all the structures observed in this study are global minima in each size. Furthermore, in the same network shapes such as chain-like and ring-like, isomers having a separated pair of H3O+ and OH are lower in energy by 3–10 kJ mol−1 than those keeping a H3O+-OH contact pair. This suggests generality of the stabilization effect in the separation of the ion-radical contact pair. “Dimer cation core” type structures as in Fig. 1a are at least 40 kJ mol−1 higher in energy than the global minimum structures. For further confirmation, IR spectra based on the most stable “dimer cation core” type structures are simulated and given in Fig. S5–7.† We found that observed spectral patterns, e.g. similarity to H+(H2O)n, are not accounted for by these simulated spectra. These results are consistent with our experimental findings and also with recent theoretical studies.833.8 Temperature (internal energy) effect
Finally, we comment on the temperature (internal energy) effect. Cluster temperature of (H2O)6+ in this study is estimated to be about 180 K by comparing the free OH band widths with those in the IR spectra of temperature-controlled H+(H2O)6 reported by Wang et al.68 To cool down clusters, we carried out messenger method (Ar-tagging).65,72,79 The internal energy of the Ar-tagged clusters(H2O)n+·Ar must be lower than the binding energy with Ar (a few hundred cm−1) because clusters with a higher internal energy should dissociate spontaneously and do not survive until the observation. Temperature of the Ar-tagged ion is estimated to be several tens of Kelvins. The obtained spectrum of (H2O)6+·Ar (Fig. 8a) shows the very similar spectral features to those of bare (H2O)6+ (Fig. 8b), except for the narrower band widths in (H2O)6+·Ar, implying that the temperature (internal energy) effect on cluster structures is not remarkable in the temperature range below ∼180 K. On the other hand, to heat the clusters, we changed the ion source to the “warmer” one with the jet expansion at much lower stagnation pressure of 0.3 MPa (Ar carrier) (details of this warmer ion source are given in the ESI†). Here, “warmer” means that the lower stagnation pressure should reduce the degree of collisional cooling (final relative temperature reflects the degree of evaporative cooling as well as collisional cooling and then it is not clear in this study). Fig. 9 compares typical mass distributions of our cold (as described in Experimental) and “warmer” ion sources. In the “warmer” ion source, we could not observe (H2O)n+ but only H+(H2O)n was observed. This cooling condition dependence of the (H2O)n+ yield is consistent with the previous mass spectrometric studies by Shinohara et al. and Jongma et al., in which they suggested rapid cooling processes after the ionization are necessary to produce (H2O)n+.29,30 The absence of (H2O)n+ in the “warmer” condition indicates that the OH radical is easily ejected by thermal evaporation, resulting in production of H+(H2O)n−x. These results are consistent with the present IR spectroscopic findings for (H2O)n+ that the OH radical is separated from the protonated site and is bound by a weaker H-bond than water–water H-bonds.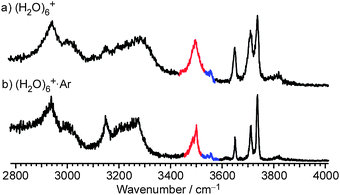 | ||
| Fig. 8 Comparison of IR spectra obtained with and without argon-tagging. (a) IR spectrum of (H2O)6+·Ar (colder). (b) IR spectrum of (H2O)6+. | ||
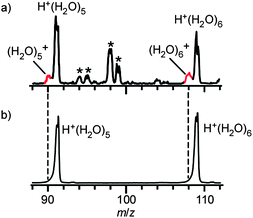 | ||
| Fig. 9 Comparison of cluster ion distributions obtained with (a) “colder” (higher stagnation pressure, 5 MPa) clusterion source and (b) “warmer” (lower stagnation pressure, 0.3 MPa). See text and the ESI† for details of each ion source. The bands indicated by the asterisk are attributed to (H2O)n+·Arm or H+(H2O)n·Arm. | ||
4. Conclusions
To investigate detailed structures of cationized water networks, we reported the IR photodissociation spectra of the watercluster radical cations (H2O)n+ (n = 3–11) in the OH stretch region. The spectral similarity to H+(H2O)n and the presence of the OH radical stretch band show that nominal watercluster cations (H2O)n+ form proton-transferred type clustersH+(H2O)n−1(OH) and also that shapes of the H-bond network are quite similar to those of H+(H2O)n. Because an OH radical is a weaker hydrogen bond acceptor than a water molecule, the first solvation shell of the protonated site is preferentially filled with water. As a result, the OH radical is separated from the protonated site by at least one water molecule in the n ≥ 5 clusters. Generality of this ion-radical separation is confirmed in both the colder and the warmer ion source conditions. According to these findings, structures of (H2O)n+ can be constructed from the structures of H+(H2O)n by substituting one of the water molecules away from the protonated site with an OH radical. Reactivity of water exposed to ionizing radiation should depend on H-bond network structures around the created radicals. We hope findings in this study would be helpful to understand detailed processes of ionizing radiation-initiated reactions in liquid water and aqueous solutions.Acknowledgements
We thank Dr Y. Matsuda, Dr T. Maeyama, and Professor N. Mikami in Tohoku University for stimulating discussions. This study was supported by the Grant-in-Aid for Scientific Research (Project No. 19056001 from MEXT Japan, and Nos. 20·5015, and 2235001 from JSPS). K.M. is supported by JSPS Research Fellowships for Young Scientists. Part of the calculations were performed at the Research Center for Computational Science, Okazaki (Japan).Notes and references
- I. G. Draganic and Z. D. Draganic, The radiation chemistry of water, ACADEMIC PRESS, New York, 1971 Search PubMed.
- B. C. Garrett, D. A. Dixon, D. M. Camaioni, D. M. Chipman, M. A. Johnson, C. D. Jonah, G. A. Kimmel, J. H. Miller, T. N. Rescigno, P. J. Rossky, S. S. Xantheas, S. D. Colson, A. H. Laufer, D. Ray, P. F. Barbara, D. M. Bartels, K. H. Becker, K. H. Bowen, S. E. Bradforth, I. Carmichael, J. V. Coe, L. R. Corrales, J. P. Cowin, M. Dupuis, K. B. Eisenthal, J. A. Franz, M. S. Gutowski, K. D. Jordan, B. D. Kay, J. A. LaVerne, S. V. Lymar, T. E. Madey, C. W. McCurdy, D. Meisel, S. Mukamel, A. R. Nilsson, T. M. Orlando, N. G. Petrik, S. M. Pimblott, J. R. Rustad, G. K. Schenter, S. J. Singer, A. Tokmakoff, L.-S. Wang and T. S. Zwier, Chem. Rev., 2005, 105, 355–390 CrossRef CAS.
- S. Lehnert, Biomolecular Action of Ionizing Radiation, Taylor & Francis Group, New York, 2008 Search PubMed.
- J. F. Ward, Prog. Nucleic Acid Res. Mol. Biol., 1988, 35, 95–125 Search PubMed.
- S. G. Swarts, M. D. Sevilla, D. Becker, C. J. Tokar and K. T. Wheeler, Radiat. Res., 1992, 129, 333–344 CAS.
- J. Cadet, T. Douki, D. Gasparutto and J.-L. Ravanat, Mutat. Res., 2003, 531, 5–23 CrossRef CAS.
- C.-R. Wang, J. Nguyen and Q.-B. Lu, J. Am. Chem. Soc., 2009, 131, 11320–11322 CrossRef CAS.
- C. Silva, P. K. Walhout, K. Yokoyama and P. F. Barbara, Phys. Rev. Lett., 1998, 80, 1086 CrossRef CAS.
- Y. Gauduel, S. Pommeret, A. Migus and A. Antonetti, Chem. Phys., 1990, 149, 1–10 CrossRef CAS.
- C. L. Thomsen, D. Madsen, S. R. Keiding, J. Thogersen and O. Christiansen, J. Chem. Phys., 1999, 110, 3453–3462 CrossRef.
- A. Migus, Y. Gauduel, J. L. Martin and A. Antonetti, Phys. Rev. Lett., 1987, 58, 1559 CrossRef CAS.
- V. Buch, S. Bauerecker, J. P. Devlin, U. Buck and J. K. Kazimirski, Int. Rev. Phys. Chem., 2004, 23, 375–433 CrossRef CAS.
- U. Buck and F. Huisken, Chem. Rev., 2000, 100, 3863–3890 CrossRef CAS.
- K. Mizuse, T. Hamashima and A. Fujii, J. Phys. Chem. A, 2009, 113, 12134–12141 CrossRef CAS.
- J. V. Coe, G. H. Lee, J. G. Eaton, S. T. Arnold, H. W. Sarkas, K. H. Bowen, C. Ludewigt, H. Haberland and D. R. Worsnop, J. Chem. Phys., 1990, 92, 3980–3982 CrossRef CAS.
- J. V. Coe, Int. Rev. Phys. Chem., 2001, 20, 33–58 CrossRef CAS.
- A. E. Bragg, J. R. R. Verlet, A. Kammrath, O. Cheshnovsky and D. M. Neumark, Science, 2004, 306, 669–671 CrossRef CAS.
- J. R. R. Verlet, A. E. Bragg, A. Kammrath, O. Cheshnovsky and D. M. Neumark, Science, 2005, 307, 93–96 CrossRef CAS.
- A. E. Bragg, J. R. R. Verlet, A. Kammrath, O. Cheshnovsky and D. M. Neumark, J. Am. Chem. Soc., 2005, 127, 15283–15295 CrossRef CAS.
- D. M. Neumark, Mol. Phys., 2008, 106, 2183–2197 CrossRef CAS.
- O. T. Ehrler and D. M. Neumark, Acc. Chem. Res., 2009, 42, 769–777 CrossRef CAS.
- T. Maeyama, T. Tsumura, A. Fujii and N. Mikami, Chem. Phys. Lett., 1997, 264, 292–296 CrossRef CAS.
- P. Ayotte and M. A. Johnson, J. Chem. Phys., 1997, 106, 811–814 CrossRef CAS.
- N. I. Hammer, J. W. Shin, J. M. Headrick, E. G. Diken, J. R. Roscioli, G. H. Weddle and M. A. Johnson, Science, 2004, 306, 675–679 CrossRef CAS.
- N. I. Hammer, J. R. Roscioli, J. C. Bopp, J. M. Headrick and M. A. Johnson, J. Chem. Phys., 2005, 123, 244311 CrossRef.
- K. R. Asmis, G. Santambrogio, J. Zhou, E. Garand, J. Headrick, D. Goebbert, M. A. Johnson and D. M. Neumark, J. Chem. Phys., 2007, 126, 191105 CrossRef.
- L. A. Posey, M. J. DeLuca, P. J. Campagnola and M. A. Johnson, J. Phys. Chem., 1989, 93, 1178–1181 CrossRef CAS.
- O. P. Balaj, C.-K. Siu, I. Balteanu, M. K. Beyer and V. E. Bondybey, Int. J. Mass Spectrom., 2004, 238, 65–74 Search PubMed.
- H. Shinohara, N. Nishi and N. Washida, J. Chem. Phys., 1986, 84, 5561–5567 CrossRef CAS.
- R. T. Jongma, Y. Huang, S. Shi and A. M. Wodtke, J. Phys. Chem. A, 1998, 102, 8847–8854 CrossRef CAS.
- L. Angel and A. J. Stace, Chem. Phys. Lett., 2001, 345, 277–281 CrossRef CAS.
- S. Yamaguchi, S. Kudoh, Y. Kawai, Y. Okada, T. Orii and K. Takeuchi, Chem. Phys. Lett., 2003, 377, 37–42 CrossRef CAS.
- S. Tomoda and K. Kimura, Chem. Phys., 1983, 82, 215–227 CrossRef CAS.
- K. Norwood, A. Ali and C. Y. Ng, J. Chem. Phys., 1991, 95, 8029–8037 CrossRef CAS.
- S. P. de Visser, L. J. de Koning and N. M. M. Nibbering, J. Phys. Chem., 1995, 99, 15444–15447 CrossRef CAS.
- R. N. Barnett and U. Landman, J. Phys. Chem., 1995, 99, 17305–17310 CrossRef CAS.
- R. N. Barnett and U. Landman, J. Phys. Chem. A, 1997, 101, 164–169 CrossRef CAS.
- Y. V. Novakovskaya and N. F. Stepanov, Int. J. Quantum Chem., 1997, 61, 981–990 CrossRef CAS.
- Y. V. Novakovskaya and N. F. Stepanov, J. Phys. Chem. A, 1999, 103, 3285–3288 CrossRef CAS.
- H. Tachikawa, J. Phys. Chem. A, 2002, 106, 6915–6921 CrossRef CAS.
- H. Tachikawa, J. Phys. Chem. A, 2004, 108, 7853–7862 CrossRef CAS.
- A. Furuhama, M. Dupuis and K. Hirao, J. Chem. Phys., 2006, 124, 164310 CrossRef CAS.
- I. B. Muller and L. S. Cederbaum, J. Chem. Phys., 2006, 125, 204305 CrossRef.
- L. Belau, K. R. Wilson, S. R. Leone and M. Ahmed, J. Phys. Chem. A, 2007, 111, 10075–10083 CrossRef CAS.
- Y. Novakovskaya, Russ. J. Phys. Chem. A, 2007, 81, 216–224 Search PubMed.
- Y. Novakovskaya, Russ. J. Phys. Chem. A, 2007, 81, 225–234 Search PubMed.
- Y. V. Novakovskaya, Int. J. Quantum Chem., 2007, 107, 2763–2780 CrossRef CAS.
- S. Yang, S. M. Brereton, S. Nandhra, A. M. Ellis, B. Shang, L.-F. Yuan and J. Yang, J. Chem. Phys., 2007, 127, 134303 CrossRef.
- A. Furuhama, M. Dupuis and K. Hirao, Phys. Chem. Chem. Phys., 2008, 10, 2033–2042 RSC.
- A. Kumar, M. Kołaski, H. M. Lee and K. S. Kim, J. Phys. Chem. A, 2008, 112, 5502–5508 CrossRef CAS.
- P. A. Pieniazek, J. VandeVondele, P. Jungwirth, A. I. Krylov and S. E. Bradforth, J. Phys. Chem. A, 2008, 112, 6159–6170 CrossRef CAS.
- Q. Cheng, F. A. Evangelista, A. C. Simmonett, Y. Yamaguchi and H. F. Schaefer, J. Phys. Chem. A, 2009, 113, 13779–13789 CrossRef CAS.
- H. M. Lee and K. S. Kim, J. Chem. Theory Comput., 2009, 5, 976–981 CrossRef CAS.
- G. Periyasamy, R. D. Levine and F. Remacle, Chem. Phys., 2009, 366, 129–138 CrossRef CAS.
- P. A. Pieniazek, E. J. Sundstrom, S. E. Bradforth and A. I. Krylov, J. Phys. Chem. A, 2009, 113, 8141–8141 CrossRef CAS.
- T. Jahnke, H. Sann, T. Havermeier, K. Kreidi, C. Stuck, M. Meckel, M. Schoffler, N. Neumann, R. Wallauer, S. Voss, A. Czasch, O. Jagutzki, A. Malakzadeh, F. Afaneh, T. Weber, H. Schmidt-Bocking and R. Dorner, Nat. Phys., 2010, 6, 139–142 CrossRef CAS.
- E. Kamarchik, O. Kostko, J. M. Bowman, M. Ahmed and A. I. Krylov, J. Chem. Phys., 2010, 132, 194311 CrossRef.
- M. Mucke, M. Braune, S. Barth, M. Forstel, T. Lischke, V. Ulrich, T. Arion, U. Becker, A. Bradshaw and U. Hergenhahn, Nat. Phys., 2010, 6, 143–146 CrossRef CAS.
- S. Barth, M. Ončaćk, V. Ulrich, M. Mucke, T. Lischke, P. Slavićček and U. Hergenhahn, J. Phys. Chem. A, 2009, 113, 13519–13527 CrossRef CAS.
- M. Sodupe, J. Bertran, L. Rodriguez-Santiago and E. J. Baerends, J. Phys. Chem. A, 1998, 103, 166–170.
- G. H. Gardenier, M. A. Johnson and A. B. McCoy, J. Phys. Chem. A, 2009, 113, 4772–4779 CrossRef CAS.
- Y. Matsuda, N. Mikami and A. Fujii, Phys. Chem. Chem. Phys., 2009, 11, 1279–1290 RSC.
- M. Hachiya, Y. Matsuda, K.-i. Suhara, N. Mikami and A. Fujii, J. Chem. Phys., 2008, 129, 094306 CrossRef.
- L. I. Yeh, M. Okumura, J. D. Myers, J. M. Price and Y. T. Lee, J. Chem. Phys., 1989, 91, 7319–7330 CrossRef CAS.
- M. Okumura, L. I. Yeh, J. D. Myers and Y. T. Lee, J. Phys. Chem., 1990, 94, 3416–3427 CrossRef CAS.
- Y. S. Wang, J. C. Jiang, C. L. Cheng, S. H. Lin, Y. T. Lee and H. C. Chang, J. Chem. Phys., 1997, 107, 9695–9698 CrossRef CAS.
- J. C. Jiang, Y. S. Wang, H. C. Chang, S. H. Lin, Y. T. Lee and G. Niedner-Schatteburg, J. Am. Chem. Soc., 2000, 122, 1398–1410 CrossRef CAS.
- Y. S. Wang, C. H. Tsai, Y. T. Lee, H. C. Chang, J. C. Jiang, O. Asvany, S. Schlemmer and D. Gerlich, J. Phys. Chem. A, 2003, 107, 4217–4225 CrossRef CAS.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Science, 2004, 304, 1134–1137 CrossRef CAS.
- J. W. Shin, N. I. Hammer, E. G. Diken, M. A. Johnson, R. S. Walters, T. D. Jaeger, M. A. Duncan, R. A. Christie and K. D. Jordan, Science, 2004, 304, 1137–1140 CrossRef CAS.
- H. C. Chang, C. C. Wu and J. L. Kuo, Int. Rev. Phys. Chem., 2005, 24, 553–578 CrossRef CAS.
- J. M. Headrick, E. G. Diken, R. S. Walters, N. I. Hammer, R. A. Christie, J. Cui, E. M. Myshakin, M. A. Duncan, M. A. Johnson and K. D. Jordan, Science, 2005, 308, 1765–1769 CrossRef CAS.
- C. K. Lin, C. C. Wu, Y. S. Wang, Y. T. Lee, H. C. Chang, J. L. Kuo and M. L. Klein, Phys. Chem. Chem. Phys., 2005, 7, 938–944 RSC.
- C.-C. Wu, C.-K. Lin, H.-C. Chang, J.-C. Jiang, J.-L. Kuo and M. L. Klein, J. Chem. Phys., 2005, 122, 074315 CrossRef.
- S. Karthikeyan, M. Park, I. Shin and K. S. Kim, J. Phys. Chem. A, 2008, 112, 10120–10124 CrossRef CAS.
- Q. C. Nguyen, Y.-S. Ong and J.-L. Kuo, J. Chem. Theory Comput., 2009, 5, 2629–2639 CrossRef CAS.
- K. Mizuse, A. Fujii and N. Mikami, J. Chem. Phys., 2007, 126, 231101 CrossRef.
- G. E. Douberly, A. M. Ricks and M. A. Duncan, J. Phys. Chem. A, 2009, 113, 8449–8453 CrossRef CAS.
- G. E. Douberly, R. S. Walters, J. Cui, K. D. Jordan and M. A. Duncan, J. Phys. Chem. A, 2010, 114, 4570–4579 CrossRef CAS.
- S. Maeda and K. Ohno, J. Phys. Chem. A, 2007, 111, 4527–4534 CrossRef CAS.
- Y. Luo, S. Maeda and K. Ohno, J. Phys. Chem. A, 2007, 111, 10732–10737 CrossRef CAS.
- Y. Luo, S. Maeda and K. Ohno, J. Comput. Chem., 2009, 30, 952–961 CrossRef CAS.
- E. Livshits, R. S. Granot and R. Baer, J. Phys. Chem. A, 2010 DOI:10.1021/jp1057572.
- K. Mizuse, N. Mikami and A. Fujii, Angew. Chem., Int. Ed., 2010, 49, 10119–10122 CrossRef CAS.
- L. Ma, K. Majer, F. Chirot and B. von Issendorff, J. Chem. Phys., 2009, 131, 144303 CrossRef.
- C. Y. Ng, D. J. Trevor, P. W. Tiedemann, S. T. Ceyer, P. L. Kronebusch, B. H. Mahan and Y. T. Lee, J. Chem. Phys., 1977, 67, 4235–4237 CrossRef CAS.
- U. Even, J. Jortner, D. Noy, N. Lavie and C. Cossart-Magos, J. Chem. Phys., 2000, 112, 8068–8071 CrossRef CAS.
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. Montgomery, J. A., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- P. Flukiger, H. P. Luthi, S. Portmann and J. Weber, MOLEKEL 4.3, Swiss Center for Scientific Computing, Manno (Switzerland), 2000–2002 Search PubMed.
- K. Ohno and S. Maeda, Chem. Phys. Lett., 2004, 384, 277–282 CrossRef CAS.
- S. Maeda and K. Ohno, J. Phys. Chem. A, 2005, 109, 5742–5753 CrossRef CAS.
- K. Ohno and S. Maeda, J. Phys. Chem. A, 2006, 110, 8933–8941 CrossRef CAS.
- Detailed descriptions of network shapes and spectral analyses for H+(H2O) n have been reported elsewhere, and the similar discussion is probably applicable to (H2O) n+. Briefly, bands are categorized to four band series, shown as dotted lines in Fig. 2. From the low frequency side, symmetric stretch of 1-coordinated (1-coord) water, free OH stretch of 3-coord water, free OH stretch of 2-coord water, and antisymmetric stretch of 1-coord water, respectively. With increasing cluster size, bands of higher coordination numbers become stronger and 1-coord water bands fade out, corresponding to structural development from chain (n ≤ 3) to tree (4 ≤ n ≤ ∼7), branched ring (∼7 ≤ n ≤ ∼10), and closed net (multiple ring) structures (n ≥ ∼10).
- K. Tsuji and K. Shibuya, J. Phys. Chem. A, 2009, 113, 9945–9951 CrossRef CAS.
- R. C. Herman and G. A. Hornbeck, Astrophys. J., 1953, 118, 214–227 CAS.
- Y. Maréchal, The Hydrogen Bond and the Water Molecule, ELSEVIER, Amsterdam, 2007 Search PubMed.
- J. R. Roscioli, L. R. McCunn and M. A. Johnson, Science, 2007, 316, 249–254 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectrum of our ion source, fragment mass spectrum of (H2O)5+, free energy calculations and relative populations for (H2O)5+, simulated IR spectra for “dimer cation core” type isomers, parameters used in the GRRM program, and details of the “warmer” ion source. See DOI: 10.1039/c0sc00604a |
| This journal is © The Royal Society of Chemistry 2011 |