Asymmetric catalytic reactions by NbO-type chiral metal–organic frameworks†‡
Kyung Seok
Jeong
a,
Yong Bok
Go
b,
Sung Min
Shin
a,
Suk Joong
Lee
a,
Jaheon
Kim
c,
Omar M.
Yaghi
b and
Nakcheol
Jeong
*ad
aDepartment of Chemistry, Korea University, Seoul, 136–701, Korea. E-mail: njeong@korea.ac.kr
bDepartment of Chemistry and Biochemistry, Center for Reticular Chemistry at the California NanoSystems Institute, University of California, Los Angeles, CA 90095-1569, USA
cDepartment of Chemistry, Soongsil University, Seoul, 156-743, Korea
dKorea Basic Science Institute, Seoul, 136-713, Korea
First published on 25th February 2011
Abstract
Chiral metal–organic frameworks (MOFs) constitute a unique class of multifunctional hybrid materials and are envisioned as a versatile tool for various enantioselective applications, including the separation of optical isomers and the promotion of catalytic enantioselective reactions. Despite some pioneering works on catalytic enantioselective reactions promoted by chiral MOFs, there is still a need for practical catalysts and many fundamental issues must be answered; such as pin-pointing the site of the reaction and expedition of the reaction rate to the level of that in homogeneous media. We have designed and synthesized a chiral metal–organic framework, (S)-KUMOF-1 (Cu2(S)-1)2(H2O)2, 1 = 2,2′-dihydroxy-6,6′-dimethyl(1,1′-biphenyl)-4,4′-dicarboxylate) of which a non-interpenetrating NbO type framework provides a spacious pore (2 × 2 × 2 nm3) and is equipped with potential catalytic sites exposed into the pore. Since the functional group on the organic links, biphenols in this MOF, can be modified further on demand, this MOF can serve as a platform for new heterogeneous catalysis. Two reactions, the carbonyl-ene reaction with modified MOF after replacement of the protons on biphenol on the organic links with Zn(II) and the hetero-Diels–Alder reaction with Ti(IV), respectively, were studied. In this manoeuver, we observed that the reaction occurs entirely inside the pores and the reaction rate of the heterogeneous reaction by this specific MOF is comparable to that of its homogeneous counterpart. In addition, it is also observed that the enantioselectivities are significantly improved by extra steric bias provided from the frames of the MOF. These observations reinforce the legitimacy of the strategy of using a chiral MOF as a highly enantioselective heterogeneous catalyst.
Introduction
Metal–organic frameworks (MOFs) are constructed using a modular approach from organic links and metal ion units, with control of both their architecture and chemical functionality.1 These frameworks have many applications in the separation and the adsorption of chemicals that exploit the intrinsic microporosity of their crystal structure.2 In addition to this feature, these materials are envisioned as a compilation of enzyme active sites if the pores bear the proper functionality.3Chiral metal–organic frameworks (MOFs), which can be assembled using a modular approach with chiral organic links and metal ions, are of interest for applications in enantioselective separation and catalysis being especially important for the chemical and pharmaceutical industries. In fact, the catalytically active chiral organic zeolite analogues and their applications in enantioselective reactions have appeared in the literature, but there has been limited success in terms of the catalytic turnover and stereoselectivity.4 This limitation on catalytic utility has two main causes; the lack of void space, which is required for practical applications, and/or the chirality attenuation during the assembly of MOF.
Some of the previously reported chiral MOFs were obtained as multiply interpenetrated frameworks4e or a single framework but with large appendages on the link4d,h to minimize the void space and the free inner space of the pore. And thus, only catalytic sites near the surface of the materials are assumed to be effective for the reactions. In recent years, chiral MOFs having large free void space have appeared in the literature.4a,b With these, it is reasonably assumed that the reactions occur not only at the surface, but also inside pores. However, the evidence is still circumstantial.5
In addition, the chirality attenuation that is associated with the assembly of the chiral units during the formation of the MOF structure is another serious challenge. In this case, as seen from cyclodextrins,6 the guests in the pore do not fully experience chiral environment to provide high stereoselectivity. Therefore, even with a large enough void space, enantioselective reaction by those MOFs suffer from negligible or marginal enantioselectivity. 4f,i
In this report, a chiral MOF, (S)-KUMOF-1, having a large void space accommodating the whole molecules without severe diffusion impedance was synthesized.7,8 Its organic linker has been devised to maintain local chirality with compact functional groups, which would contribute to produce a catalytic site. With proper modification of the appended hydroxyl groups on the organic link, both the stoichiometric carbonyl-ene reaction9 and the catalytic hetero-Diels–Alder reaction10 by using Zn(II)- or Ti(VI)-decorated chiral MOFs, respectively, were successfully exercised providing excellent stereoselectivities. Additionally, the stoichiometric carbonyl-ene reaction shows particular but clear evidence that the reaction occurs inside the MOF pore.5
Experimental section
Synthesis of [Cu2((S)-1)2(H2O)2] ((S)-KUMOF-1)
Cu(NO3)2·3H2O (7.2 mg, 0.030 mmol) and (S)-1H2 (9.0 mg, 0.030 mmol) in a solvent mixture of DEF/MeOH (1.5/1.5 mL) were placed in a 4 mL vial. N,N′-dimethylaniline (1 mL) was placed in a 20 mL vial. The 4 mL vial was placed in the center of the 20 mL vial, which was capped tightly. The system was heated at 65 °C for 1 day, blue cubic crystals were obtained in 35% yield based on the used ligand. The blue crystals in dark blue solution were rinsed with a solvent mixture of DEF/MeOH (3/3 mL) three times. Elementary analysis: (%) As the free guest molecules were evaporated before the conduction of the elemental analysis. calcd. for (S)-KUMOF-1 [Cu2((S)-1)2]·(H2O)2 = C32H28O14Cu2: C, 50.33; H, 3.70; found: C, 50.38; H, 3.75. IR (KBr): 3407 (br), 2924 (w), 1655 (s), 1559 (s), 1414 (s), 1296 (w), 1248 (w), 1165 (w), 1103 (m), 1041 (m), 1000 (m), 882 (w), 793 (m), 772 (m), 662 (w) cm−1X-Ray crystallography
(S)-KUMOF-1 is cubic: I432 (No. 211), a = b = c = 30.1892(2) Å, V = 27514.1(3) Å3, Z = 6. Total reflections, 53379; independent reflections, 3874 [Rint = 0.0471]; parameters, 68; after SQUEEZE, R1 = 0.0613, wR2 = 0.1891 (I > 2σ(I)); Dc = 0.277 g cm−3 for the framework only. CCDC reference no. 775002.11Preparation of Zn/(S)-KUMOF-1
To a solution of solvent exchanged (S)-KUMOF-1 (102.0 mg, 0.27 mmol) in dichloromethane (2 mL) was added dimethylzinc (0.4 mL, 2 M in dichloromethane, 0.81 mmol) at −78 °C and the solution was stirred for 3 h at this temperature. Zn/(S)-KUMOF-1 was washed with cold dichloromethane several times.Carbonyl-ene reaction of 2 by Zn/(S)-KUMOF-1
To a solution of the Zn/(S)-KUMOF-1 in fresh dichloromethane (2 mL) a mixture of 2 (15.0 mg, 0.089 mmol) in dichloromethane (0.1 mL) was added at −78 °C. The solution was warmed to 0 °C and stirred for 3 h at this temperature. To the reaction mixture was added an aqueous solution of 6 N HCl (3 mL) and the resultant mixture was filtered through Celite. The filtrate was concentrated in vacuo and the residue was purified by flash chromatography (n-hexane–ethyl acetate 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 3 (13.8 mg, 92% yield) Rf 0.43 (n-Hex:EA = 10:1)
:1) to give 3 (13.8 mg, 92% yield) Rf 0.43 (n-Hex:EA = 10:1)
Preparation of Mosher ester and determination of stereoselectivity
Compound 3 was treated with α-methoxy-α-trifluoromethylphenylacetic acid (MTPA) and pyridine in dichloromethane to give 4, as a diastereomeric mixture, and the resultant mixture was subjected to gas chromatography for determining optical purity.11Preparation of Ti/(S)-KUMOF-1
A mixture of (S)-KUMOF-1 (24.0 mg, 0.063 mmol), Ti(O-iPr)4 (0.2 mL, 1 M in toluene, 0.20 mmol) in toluene (1.5 mL) was stirred for 5 h at 25 °C. Ti/(S)-KUMOF-1 was washed with cold toluene several times.Hetero-Diels–Alder reaction of 6 and 7
The Ti/(S)-KUMOF-1 in fresh toluene (1.5 mL) was cooled to 0 °C and then Danishefsky's diene 6 (41 μL, 0.21 mmol), benzaldehyde 7 (21 μL, 0.21 mmol) was added portionwise. The mixture was stirred at 0 °C for 3 days and then treated with 5 drops of trifluoroacetic acid. After the mixture was stirred at 0 °C for 15 min, a saturated aqueous solution of NaHCO3 (1.5 mL) was added. The mixture were stirred for 10 min and then filtered through a plug of Celite. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 × 1 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (SiO2, EtOAc–petroleum ether 1:4, Rf: 0.5) to yield (R)-2-phenyl-2,3-dihydro-4H-pyran-4-one (9) (30.7 mg, 0.18 mmol, 84% yield) as a yellow oil. (See ESI for details.‡)
Results and discussion
Rationale of the ligand design
This study was carried out with (S)-1H2, enantiopure (S)-2,2′-dihydroxy-6,6′-dimethyl-(1,1′-biphenyl)-4,4′-dicarboxylic acid or its antipode (R)-1H2 (Fig. 1a), which were synthesized from a previously reported intermediate (S)-4,4'-dimethoxy-6,6′-dimethyl(1,1′-biphenyl)-2,2′-dicarboxylic acid12 (Scheme S1 in ESI‡). The main idea lying at the selection of the candidate linker was to embody chirality with a substituent as small as possible. A methyl group was chosen as the substituent because it is large enough to prevent free rotation along the axes that connect two phenyl groups, creating the chirality, but it is small enough not to occupy too much of the pore space. Additionally, the link is nearly symmetric because the size of the methyl group is comparable to that of the hydroxyl group. Then, the hydroxyl groups can be used as ligating sites for the potential catalytic metal centers.![Crystal structure of a porous (S)-KUMOF-1; (a) the building blocks: Cu(ii) paddle-wheel unit and an organic linker, (S)-1H2; (b) NbO-type framework structure of (S)-KUMOF-1 (one unit cell) is shown with disordered methyl and hydroxyl groups; (c) channels in (S)-KUMOF-1 are present along the crystallographic [111] direction (8 unit cells).](/image/article/2011/SC/c0sc00582g/c0sc00582g-f1.gif) | ||
| Fig. 1 Crystal structure of a porous (S)-KUMOF-1; (a) the building blocks: Cu(II) paddle-wheel unit and an organic linker, (S)-1H2; (b) NbO-type framework structure of (S)-KUMOF-1 (one unit cell) is shown with disordered methyl and hydroxyl groups; (c) channels in (S)-KUMOF-1 are present along the crystallographic [111] direction (8 unit cells). | ||
Synthesis and characterization of (S)-KUMOF-1
In a mixed solvent (MeOH: DEF = 1: 1 in v/v), (S)-1H2 and Cu(NO3)2 were dissolved, and into the mixture N,N-dimethylaniline vapor was diffused at 65 °C for 1 day to produce blue block MOF crystals. Based on elemental analysis, thermogravimetric analysis, 1H-NMR measurement of acid-digested sample, and X-ray single crystal diffraction analysis, the crystal was formulated as Cu2(H2O)2(S)-1·(DEF)7.6(MeOH)9.6 (S)-KUMOF-1.11 The bulk phase purity was confirmed by powder X-ray diffraction (PXRD). (Figure S4 in ESI‡) The single-crystal X-ray diffraction analysis of (S)-KUMOF-1 reveals that it crystallizes into a cubic chiral space group, I432. However, the overall structure apparently looks achiral with Im(-3)m space group symmetry because both hydroxyl and methyl groups in (S)-1 are disordered with equal probabilities at same positions. This means that (S)-1s are arranged with random orientations in the framework. Nevertheless, the framework contains a homochiral (S)-1 linker, and thus (S)-KUMOF-1 must keep chirality locally on its near symmetric framework.Two Cu(II) ions are held by four carboxylate groups of (S)-1s to form a paddle-wheel secondary building unit (SBU) (Fig. 1a).11 Because of the steric repulsion between the 2,2′-dihydroxy and 6,6′-dimethyl groups of each side, two phenyls in (S)-1 are almost orthogonal to each other with a dihedral angle of 84.5°.
In turn, the two paddle-wheel clusters at each end of biphenyl are perpendicular to each other at a distance of 15.62 Å, and this connectivity pattern led to an NbO-type network topology that was first exemplified by MOF-101 (Cu2(o-Br-BDC)2(H2O)2).13 Contrary to previously reported expanded NbO-type MOFs containing similar biphenyl dicarboxylate linkers,13a (S)-KUMOF-1 has a non-interpenetrating framework, which resulted in a large void space; the interatomic distances among three sets of opposite water oxygen atoms are 23.16 Å, and so a 2 × 2 × 2 nm3 imaginary cube can reside without touching van der Waals surfaces of the coordinating water molecules (Fig. 1b). This is one of the largest void spaces available among the known chiral MOFs. (S)-KUMOF-1 also provides a large hexagonal channel with a free aperture diameter of 1.4 nm along the [111] direction (Fig. 1c). As a result, it has a very low framework density of 0.28 g cm−3 and 87% the unit cell volume can be occupied by guest molecules.
Since (S)-KUMOF-1 exhibited severe irreversible structural deformations under reduced pressure, the occluded solvents in the MOF crystals were replaced by continuous washing with relatively inert solvents, such as toluene, THF, and/or dichloromethane. The XRPD study confirmed that the MOF structure was intact after solvent exchange (Fig. S4 in ESI‡).
A stoichiometric carbonyl-ene reaction by (S)-KUMOF-1: A useful probe for demonstrating the reactions inside pores
The Lewis-acid promoted carbonyl-ene reaction represents an important transformation class. The cyclization of 3-methylgeranial 2 leading to the cyclic terpenoid analogue 3 through the Lewis-acid-promoted carbonyl-ene reaction was studied.8 In place of the zinc reagent (R)-5-Zn used in the original study, a modified (S)-KUMOF-1 with ZnMe2 (Zn/(S)-KUMOF-1; Fig. S5 in ESI for the characterization‡) was applied to the carbonyl-ene reaction as summarized in the entries 3–5 in Table 1. Product 3 was obtained as a single diastereomer as shown in Scheme 1 at 89% yield after 2 h at 0 °C from the first run with 1.5 eq of Zn/(S)-KUMOF-1 (Entry 3 in Table 1). Its optical purity was 23% ee, which was measured as a diastereomeric mixture of Mosher's ester 4. The optical purity of product 3 significantly improved as the amount of the reagent increased. With 3 eq. of Zn/(S)-KUMOF-1, the reaction produced a considerably higher chemical yield as well as a higher stereoselectivity (92% and 50% ee) after 2 h (Entry 4 in Table 1). However, additional Zn/(S)-KUMOF-1 (6 eq.) did not affect on the enantioselectivity as hoped (Entry 5 in Table 1).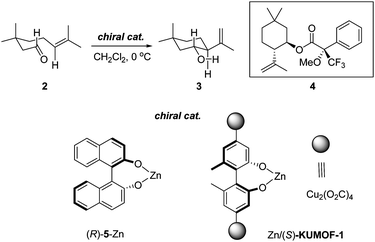 | ||
| Scheme 1 Lewis-acid-promoted carbonyl-ene reaction. | ||
| Entry | Catalyst | MOF or ligand:ZnMe2 (eq) |
t (h) | Yield of 3b (%) | de of 4c (%) |
|---|---|---|---|---|---|
| a Dimethyl zinc (0.41 mL, 2 M in dichloromethane, 0.81 mmol) was added to a solution of (S)-KUMOF-1 (102 mg, 0.27 mmol) in dichloromethane (2 mL) at −78 °C. The solution was stirred for 3 h at this temperature, and then 2 (15 mg, 0.089 mmol) in dichloromethane (0.1 mL) was added to the mixture. The solution was warmed to 0 °C and stirred for 2 h at this temperature. b Isolated yield. c Determined by GC chromatography with Mosher's ester 4. | |||||
| 1 | — | 0:9 |
2 | 0 | 0 |
| 2 | (S)-KUMOF-1 | 1.5:0 |
3.5 | 0 | 0 |
| 3a | Zn/(S)-KUMOF-1 | 1.5:4.5 |
3.5 | 89 | 23 |
| 4a | Zn/(S)-KUMOF-1 | 3:9 |
3.5 | 92 | 50 |
| 5a | Zn/(S)-KUMOF-1 | 6:18 |
3.5 | 92 | 50 |
| 6 | (R)-5-Zn | 0.2:0.2 |
2 | 20 | 37 |
| 7 | (R)-5-Zn | 1.5:1.5 |
2 | 77 | 55 |
| 8 | (R)-5-Zn | 3:3 |
2 | 86 | 88 |
| 9 | (R)-5-Zn | 6:6 |
2 | 91 | 90 |
Some critically important aspects relating to these heterogeneous reactions are as follows. First, the control experiments conducted with dimethylzinc or (S)-KUMOF-1 alone did not promote the reactions (Entries 1 and 2 in Table 1). The latter indicates that neither the bisphenol groups nor a copper ion of (S)-KUMOF-1 is responsible for this carbonyl-ene reaction. The reaction was promoted to provide 3 only by the presumed reagent, Zn/(S)-KUMOF-1,11 which was derived from (S)-KUMOF-1 by treatment with excess dimethyl zinc for 2 h at 0 °C.
Second, although the carbonyl-ene reaction was initially regarded as catalytic in principle, more than a stoichiometric amount of the Lewis acid was required in order to obtain practically acceptable results, as reported by Yamamoto.9 Even with a stoichiometric amount of the reagent, the reaction often halted half-way to completion, and the rest of the starting materials remained unreacted. Six equivalents of the Lewis acid were necessary to achieve both good yields (>90%) and high ee values (>90%) even for the reaction with (R)-5-Zn (Entries 6–9, Table 1). Thus, an excess of the chiral MOF (at least 3 eq.) was also required to complete the heterogeneous reaction.
Third, it was surprise to notice that, as the reaction progressed and even after complete disappearance of 2, product 3 did not appear; eventually, neither 2 nor 3 were detected using TLC. This indicated that the product 3 was entrapped somehow in Zn/(S)-KUMOF-1, and therefore, 3 could be obtained only after catalyst crystals was dismantled through a treatment with an aqueous HCl (6 N) solution. The product 3 has alcohol groups, and their coordinating ability to zinc ion is presumably much stronger than the carbonyl of 2. Therefore, it was suggested that 3 was bound to the Zn ion centers in the MOF even after the chemical transformation was completed, lingering there and thus blocking another reaction cycles (Fig. 2, Fig. S8 in ESI‡). This phenomenon is a kind of suicide inhibition, and explains the need for more than a stoichiometric amount of the catalyst for completing the transformation with reasonable enantioselectivity. Ironically, these features have provided a conclusive evidence to resolve the debate in many previous cases; the reaction occurs inside pores.3a,e,5 (Fig. 2 and full details of discussion can be found in ESI, Figs S9 and S10‡)
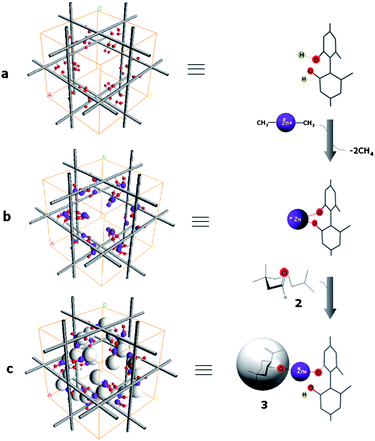 | ||
| Fig. 2 A schematic representation of the carbonyl-ene reaction inside pore: a suicide inhibition of Zn/(S)-KUMOF-1 by product 3. | ||
Fourth, the overall reaction rate for the heterogeneous MOF (completed in 3 h) was comparable to the homogeneous reaction (completed in 2 h) by (R)-5-Zn (entries 7–9 in Table 1). MOF crystals were destroyed right after all of starting material 2 disappeared in order to quench the reaction, no trace of starting material 2 was observed. It implies that once the substrates came in contact with Zn, the catalytic reaction occurred instantaneously (Fig. 2). Considering the heavy localization of the catalytic sites in the Zn/(S)-KUMOF-1, this reaction time difference is insignificant.
Although it has been reported that the diffusion of substrate inside pore of the MOF is much slower,8 it is no longer a problem for overall efficiency of this specific case.
Catalytic enantioselective hetero-Diels–Alder by (S)-KUMOF-1
While the previous reaction reflects many of the characteristic features of the reaction with chiral MOF, both the consumption of the excess amount of MOFs and their disposal after reaction make the reaction unsuitable for practical applications. Thus, for the demonstration of the efficiency of this MOF as a heterogeneous catalyst, a hetero Diels–Alder reaction between Danishefsky's diene 6 and aldehyde 7 has been chosen as a benchmark catalytic reaction (Scheme 2).10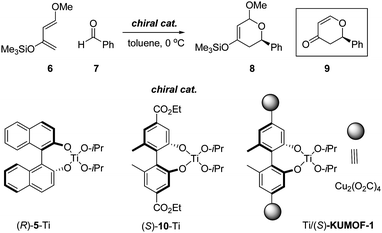 | ||
| Scheme 2 Lewis-acid-promoted hetero Diels–Alder reaction. | ||
A control experiment with 6 and 7 in the presence of (R)-5-Ti that was obtained from (R)-binaphthol ((R)-5H2) and titanium tetra-iso-propoxide (1:1.5) at 0 °C formed the corresponding product 8 after 5 days. Product 8 was characterized as 9 after acidic hydrolysis to yield it at a 72% yield. The enantioselectivity was 51% ee (Entry 8 in Table 2).11
| Entry | Catalyst | MOF or ligand:Ti(O-iPr)4 (mol%) |
t (d) | Yield of 9c (%) | ee of 9d (%) |
|---|---|---|---|---|---|
| a Reaction conditions: A mixture of (S)-KUMOF-1 (24 mg, 0.063 mmol), Ti(O-iPr)4 (0.2 mL, 1 M in toluene, 0.20 mmol) in toluene was stirred for 5 h at 25 °C. The mixture was cooled to 0 °C and then Danishefsky's diene 7 (41 μL, 0.21 mmol) and benzaldehyde 8 (22.3 mg, 0.21 mmol) were added stepwise. The mixture was stirred at 0 °C for 3–5 days and then treated with 5 drops of TFA. b The reaction was carried out at 25 °C. c Isolated yield. d Determined by LC chromatography: Chiralcel OD-H, n-hexane/iPrOH = 40/1, flow rate = 1.5 mL min−1. | |||||
| 1 | — | 0:20 |
3b | 10 | — |
| 2 | — | 0:20 |
3 | 7 | — |
| 3 | (S)-KUMOF-1 | 30:0 |
3 | NR | — |
| 4a | Ti/(S)-KUMOF-1 | 30:30 |
3 | 52 | 33 |
| 5a | Ti/(S)-KUMOF-1 | 30:45 |
5 | 77 | 48 |
| 6a | Ti/(S)-KUMOF-1 | 30:60 |
5 | 79 | 55 |
| 7a | Ti/(S)-KUMOF-1 | 30:90 |
5 | 80 | 55 |
| 8 | (R)-5-Ti | 30:90 |
5 | 72 | 51 |
| 9 | (S)-10-Ti | 30:90 |
5 | 58 | 18 |
As (S)-KUMOF-1 itself did not expedite the reaction (entry 3 in Table 2), it was treated with Ti(O-iPr)4 to make active Ti/(S)-KUMOF-1 (for the characterization, see Fig. S6 in ESI). With a catalytic amount of Ti/(S)-KUMOF-1 (30 mol%) coupled with 0.9 eq of Ti(O-iPr)4, the reaction proceeded to completion after 5 days at 0 °C, yielding 8 and then 9 eventually at 77% yield after the acidic work-up. The enantioselectivity (48% ee) is not as high as wished but is still significant (Entry 5 in Table 2). As reported for the homogeneous reaction,9 the increase in Ti(O-iPr)4 (1.5–3.0 eq) is essential to the significant improvement of the stereoselectivity. This enhancement reached a plateau (80% chemical yield and 55% ee) at a 1:2 ratio of (S)-KUMOF-1 and Ti(O-iPr)4 (Entries 4, 6 and 7 in Table 2).
Enantioselectivies of the homogeneous reaction and heterogeneous were compared. Since the enantioselectivity is dependent not only on the ligand backbone but also the steric and electronic character of substituent, the catalyst derived from ligand (S)-10-H2, which has same backbone and similar electron-withdrawing substituent at 4, 4′-position, was chosen for the fair comparison. The homogeneous reaction by this catalyst yielded 9 in a disappointing chemical yield, 58%, together with enantioselectivity, 18% ee (Entry 9 in Table 2). However, immobilization of link 1 on the MOF provided significantly improved stereoselectivity up to 55% ee with a restoring of chemical yield (80%, Entry 7 in Table 2). This improvement in enantioselectivity as well as chemical yield may be accounted for by the contribution of the MOF frameworks. In such confined area, guest substrate would experienced an exaggerated chiral environment by the frameworks, as often seen from the 2,2-disubstituted-1,1-binaphthyl skeleton equipped with bulky substituents on 3,3-position in homogeneous reaction.14 Since the copper sites in the framework do not participate in the catalytic reaction (Entry 3 in Table 2), the framework of the MOF catalyst is supposed to be remained intact even after a cycle of catalytic reaction. Indeed, as long as the reaction temperature was kept properly below 0 °C, the XRPD patterns of MOFs were virtually unchanged (Figs S13C and D in ESI‡). Thus, the repetitive use of Ti/(S)-KUMOF-1 was realized without troubles (Table S2 in ESI‡).
Conclusion
In summary, the synthesis and structure elucidation of a near mesoporous chiral MOF, (S)-KUMOF-1, with NbO topology together with a local chirality were successfully achieved. This MOF turned out to be an excellent platform of heterogeneous catalysts for enantioselective reactions after proper functionalization for endowing Lewis-acidity through metal-hydrogen exchange. It is as efficient as its homogeneous counterpart in terms of the reaction rate because the large window and void space inside of the MOF reduces the difficulties significantly associated with the diffusion impedance. In addition, from a stoichiometric carbonyl-ene reaction, conclusive evidence, for the most portion of the reaction occurred inside pores of the MOF, was obtained. The exaggerated chiral environment nearby the reaction site by the frames is also beneficial and contributes to the substantial improvement in stereoselectivity. Finally, the catalyst is recyclable as well.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant (2009-0053318) funded by the Korea government (MEST). Experiments at PLS (beam line 6b-1) were supported in part by MEST and POSTECH.Notes and references
- Representative references for MOF in general: (a) H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846 CrossRef CAS; (b) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS; (c) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC; (d) M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef CAS; (e) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
- Selected references for the separation and gas adsorption: (a) J.-R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (b) M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS.
- (a) A. Corma, H. García and F. X. Llabrési Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS. Selected references for the reaction at metal site or post-modified metal sites on the MOF: (b) S. Horike, M. Dinca, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854 CrossRef CAS; (c) P. Horcajada, S. Surblé, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820 RSC. Selected references for the reaction by the functional group on the organic links: (d) M. Banerjee, S. Das, M. Yoon, H. J. Choi, M. H. Hyun, S. M. Park, G. Geo and K. Kim, J. Am. Chem. Soc., 2009, 131, 7524 CrossRef CAS; (e) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607 CrossRef CAS; (f) T. Uemura, R. Kitaura, Y. Ohta, M. Nagaoka and S. Kitagawa, Angew. Chem., Int. Ed., 2006, 45, 4112 CrossRef CAS.
- (a) L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838 CrossRef CAS; (b) L. Ma, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390 CrossRef; (c) D. Dang, P. Wu, C. He, Z. Xie and C. Duan, J. Am. Chem. Soc., 2010, 132, 14321 CrossRef CAS; (d) K. Tanaka, S. Oda and M. Shiro, Chem. Commun., 2008, 820 RSC; (e) S.-H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563 RSC; (f) D. N. Dybtsev, A. L. Nuzhdin, H. Chun, K. P. Bryliakov, E. P. Talsi, V. P. Fedin and K. Kim, Angew. Chem., Int. Ed., 2006, 45, 916 CrossRef CAS; (g) W. Lin, J. Solid State Chem., 2005, 178, 2486 CrossRef CAS; (h) C. D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS; (i) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS.
- Especially, as long as the examined reactions are catalytic, the conclusion cannot help but be circumstantial.
- K. Kano, R. Nishiyabu, Advances in Supramolecular Chemistry, ed G. W. Gokel, Cerberus Press, Inc., South Miami, FL, , 2003, vol. 9, p39 Search PubMed.
- (a) Y. K. Park, S. B. Choi, H. Kim, K. Kim, B. H. Won, K. Choi, J. S. Choi, W. S. Ahn, N. Won, S. Kim, D. H. Jung, S. H. Choi, G. H. Kim, S. S. Cha, Y. H. Jhon, J. K. Yang and J. Kim, Angew. Chem., Int. Ed., 2007, 46, 8230 CrossRef CAS; (b) M. Jaroniec, Nature, 2006, 442, 638 CrossRef CAS; (c) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS; (d) S. Inagaki, S. Guan, S. Ohsuna and O. Terasaki, Nature, 2002, 416, 304 CrossRef CAS; (e) Q. R. Fang, G. S. Zhu, Z. Jin, Y. Y. Ji, J. W. Ye, M. Xue, H. Yang, Y. Wang and S. L. Qiu, Angew. Chem., Int. Ed., 2007, 46, 6638 CrossRef CAS; (f) H. Yang, N. Coombs and G. A. Ozin, Nature, 1997, 386, 692 CrossRef CAS.
- (a) F. Salle, H. Jobic, A. Ghoufi, P. L. Llewellyn, C. Serre, S. Bourrelly, G. Ferey and G. Maurin, Angew. Chem., Int. Ed., 2009, 48, 8335 CrossRef CAS; (b) F. Salle, H. Jobic, G. Maurin, M. M. Koza, P. L. Llewellyn, T. Devic, C. Serre and G. Ferey, Phys. Rev. Lett., 2008, 100, 245901 CrossRef CAS; (c) F. Stallmach, S. Gröger, V. Künzel, J. Kärger, O. M. Yaghi, M. Hesse and U. Mäller, Angew. Chem., Int. Ed., 2006, 45, 2123 CrossRef CAS.
- (a) S. Sakane, K. Maruoka and H. Yamamoto, Tetrahedron, 1986, 42, 2203 CrossRef CAS; (b) S. Sakane, K. Maruoka and H. Yamamoto, Tetrahedron Lett., 1985, 26, 5535 CrossRef CAS.
- (a) S. J. Danishefsky and M. P. De Ninno, Angew. Chem., Int. Ed. Engl., 1987, 26, 15 CrossRef; (b) B. Wang, X. Feng, Y. Huang, H. Liu, X. Cui and Y. Jiang, J. Org. Chem., 2002, 67, 2175 CrossRef CAS; (c) M. Anada, T. Washio, N. Shimada, S. Kitagaki, M. Nakajima, M. Shiro and S. Hashimoto, Angew. Chem., Int. Ed., 2004, 43, 2665 CrossRef CAS; (d) K. A. Jörgensen, Angew. Chem., Int. Ed., 2000, 39, 3558 CrossRef CAS; (e) D. L. Boger, S. N. Weinreb, in Hetero Diels–Alder Methodology in Organic Synthesis; Wasserman, H. H., Ed.; Academic Press: San Diego, CA, 1987; vol. 47 Search PubMed; (f) L. F. Tietze, G. Kettschau, In Stereoselective Heterocyclic Synthesis; Metz, I. P., Ed.; Springer-Verlag: Berlin, 1997; vol. 189. p. 14 Search PubMed.
- See details in the ESI‡.
- R. Holzwarth, R. Bartsch, Z. Cherkaoui and G. Solladié, Chem.–Eur. J., 2004, 10, 3931 CrossRef CAS.
- (a) H. Furukawa, J. Kim, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650 CrossRef CAS; (b) M. Eddaoudi, J. Kim, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2002, 124, 376 CrossRef CAS.
- (a) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS; (b) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Eun Lee, an inspiring teacher and mentor, on the occasion of his retirement. |
| ‡ Electronic supplementary information (ESI) available: Detailed experimental procedure of the synthesis of ligand and MOF, characterizations of MOF before and after modification, XRD data for MOFs, and experimental details for the application MOFs as catalyst. CCDC reference number 775002. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00582g |
| This journal is © The Royal Society of Chemistry 2011 |