Novel quadridentate salen type triple-decker sandwich ytterbium complexes with near infrared luminescence†‡
Peng-Fei
Yan
,
Shuo
Chen
,
Peng
Chen
,
Ju-Wen
Zhang
and
Guang-Ming
Li
*
Key Laboratory of Functional Inorganic Material Chemistry (MOE), School of Chemistry and Materials Science, Heilongjiang University, No. 74, Xuefu Road, Nangang District, Harbin, 150080, P. R. China. E-mail: gmli_2000@163.com
First published on 26th October 2010
Abstract
Unique quadridentate salen type triple-decker dinuclear sandwich ytterbium complex [Yb2L3(CH3OH)]·0.5H2O (1) and triple-decker trinuclear sandwich ytterbium complex [Yb3L3(CH3OH)2(NO3)3]·CH3OH·5H2O (2) are isolated from the reactions of Yb(OAc)3·4H2O and Yb(NO3)3·6H2O with H2L, respectively (H2L = N,N′-bis(salicylidene)-1,2-(phenylene-diamine)). Complex 2 could be harvested from the addition of complex 1 and Yb(NO3)3·6H2O. Near infrared (NIR) lanthanide luminescence and the lifetime of complexes 1 and 2 are conducted and described.
Polynuclear lanthanide complexes with distinct magnetic and luminescent properties are currently of interest because of their potential applications in the fabrication of novel materials and as probes in biological systems.1 However, the synthesis of the polynuclear lanthanide complexes is always a challenge, due to the high coordination numbers and flexible coordination geometries adopted by the Ln3+ ions. The structures of the complexes are often influenced by a variety of factors such as solvent, pH value, ionic radius and the nature of the counterions, and a wide variety of multidentate ligands have been employed in order to address these issues.2 One of the well-known multidentate ligands is the salen type ligand (Scheme 1), and numerous structures have been explored incorporated with both d- and f-block metals.3 Pioneering studies by Costes and co-workers focused on the structures and magnetic properties of the salen type 3d–4f heteronuclear complexes,4 and some lanthanide derivatives of salen type ligand have been documented.5 It is noted that only a few examples of polynuclear lanthanide derivatives of salen type ligands are known, especially for the polynuclear lanthanide derivatives with sandwich structure. Recently, Jones and co-workers have reported several interesting salen type polynuclear lanthanide derivatives with trinuclear triple-decker,6a trinuclear tetra-decker6b and pentanuclear tetra-decker structures.7
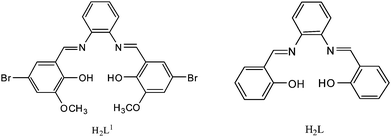 | ||
| Scheme 1 The structures of salen type ligands. | ||
However, the salen type ligands are focused only on the H2L1 (Scheme 1) which is a potential hexadentate ligand. To the best of our knowledge, the structure of polynuclear lanthanide derivatives of tetradentate salen type ligand H2L is unknown. As part of our ongoing interest in the structures and photophysical properties of salen type lanthanide complexes,8 we describe here the syntheses, structures and NIR luminescence of two novel tetradentate salen type ytterbium complexes with triple-decker dinuclear (1) and triple-decker trinuclear (2) structures. Transformation from triple-decker dinuclear 1 to triple-decker trinuclear 2 with the introduction of Yb(NO3)3·6H2O has been investigated.
Self-assembly reactions of H2L with Yb(OAc)3·4H2O and Yb(NO3)3·6H2O afford complexes 1 and 2, respectively.§X-Ray crystallographic analysis reveals that 1 has a typical triple-decker dinuclear sandwich structure (Fig. 1). The Yb13+ ion is seven-coordinated to five oxygen atoms (four from two ligands and one from the methanol molecule) and two nitrogen atoms from the bottom ligand forming a mono-capped trigonal prism geometry (Fig. S1, ESI‡). The Yb23+ ion is eight-coordinated to four oxygen atoms and four nitrogen atoms from two different ligands forming a square antiprism geometry. Yb1 and Yb2 are bridged by the O3 and O4 atoms with an Yb1⋯Yb2 distance of 3.7927 (9) Å. The bond distances of Yb–O are in the range of 2.136(6) to 2.375(5) Å, and the average distance of Yb–O bonds is 2.253 Å. Notably, the Yb–O bond distances for bridging phenolic O3 and O4 atoms (2.285(5)–2.315(5) Å) are significantly longer than those for terminal phenolic O1, O2, O5 and O6 atoms (2.136(6) to 2.245(5) Å), exhibiting weaker Yb–O bonding. The longer bond distance of Yb1–O1 than the rest is ascribed to strong H-bonding between O1 and O7 (2.782(9) Å) (Fig. S2, ESI‡), which also contribute to the stability of the complex 1. The bond distances of Yb–N are in the range of 2.407(6) to 2.541(8) Å, and the average bond distance of Yb–N is 2.466 Å. A similar bond distance is also observed for Yb–N, the bonds Yb2–N3 and Yb2–N4 from the medial ligand are longer than those of Yb2–N1, Yb2–N2, Yb1–N5 and Yb1–N6 from the other two ligands. Interestingly, the outer two ligands are twisted in a butterfly style.
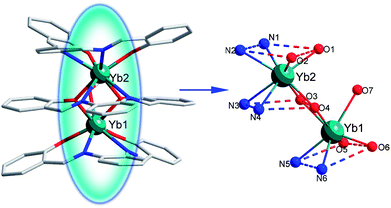 | ||
| Fig. 1 The crystal structure of 1 showing general ligand configurations and the locations of the N and O atoms. Selected bond lengths (Å): Yb1–N5 2.406(6), Yb1–N6 2.440(7), Yb1–O3 2.312(6), Yb1–O4 2.285(5), Yb1–O5 2.136(6), Yb1–O6 2.158(6), Yb1–O7 2.375(5), Yb2–O1 2.245(5), Yb2–O2 2.189(6), Yb2–O3 2.292(5), Yb2–O4 2.287(6), Yb2–N1 2.435(7), Yb2–N2 2.483(8), Yb2–N3 2.540(8), Yb2–N4 2.491(8). | ||
Complex 2 has a triple-decker trinuclear sandwich structure (Fig. 2). The coordination numbers and geometries of Yb13+ and Yb23+ ions in complex 2 are similar to those in complex 1, while the Yb33+ ion is nine-coordinated with two phenolic oxygen atoms of the ligand, one oxygen atom of one CH3OH molecule and six oxygen atoms from three bidentate NO3− anions. Therefore, Yb13+, Yb23+ and Yb33+ ions are seven-, eight-, and nine-coordinated forming a mono-capped trigonal prism, pseudo-square based antiprismatic geometry and tri-capped trigonal prism geometries, respectively. (Fig. S1, ESI‡) The Yb3+ ions are connected up by the phenolic oxygen atoms, and the distances of Yb1⋯Yb2 and Yb2⋯Yb3 are 3.768(2) Å and 3.6855(17) Å, respectively. The Yb–O bond distances are in the range of 2.129(13) to 2.504(14) Å. The average Yb–O bond distance is 2.326 Å. The Yb–N bond distances are in the range of 2.451(14)–2.496(14) Å, and the average Yb–N bond distance is 2.469 Å. A similar trend of the bond distances for Yb–O and Yb–N as that seen in complex 1 could be detected among the ligand moieties in 2. Significantly, an intramolecular H-bond could be found between O17 and O15 atoms (2.928 Å) in 2. (Fig. S3, ESI‡) It might facilitate the formation and strengthen the stability of 2.
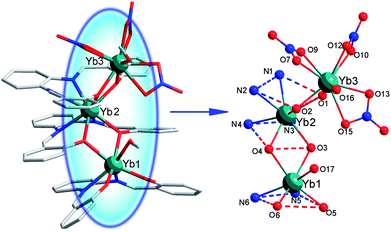 | ||
| Fig. 2 The crystal structure of 2 showing general ligand configurations and the locations of the N and O atoms. Selected bond lengths (Å): Yb1–O6 2.129(13), Yb1–O5 2.183(13), Yb1–O3 2.303(11), Yb1–O4 2.316(11), Yb1–O17 2.398(15), Yb1–N5 2.455(16), Yb1–N6 2.451(14), Yb2–O3 2.277(12), Yb2–O4 2.273(11), Yb2–O1 2.291(11), Yb2–O2 2.303(12), Yb2–N3 2.459(13), Yb2–N2 2.479(14), Yb2–N1 2.475(14), Yb2–N4 2.496(14), Yb3–O2 2.273(13), Yb3–O1 2.276(13), Yb3–O16 2.341(13), Yb3–O13 2.355(13), Yb3–O9 2.388(13), Yb3–O12 2.388(16), Yb3–O7 2.472(13), Yb3–O15 2.488(14), Yb3–O10 2.504(14). | ||
Strikingly, a close structural relationship has been found between the structure of 1 and 2, in that 2 could be seen as having been generated from the addition of 1 to Yb(NO3)3 and CH3OH. To investigate the structural relationship of 1 and 2, finely ground powder of 1 and Yb(NO3)3·6H2O were added into a solution (5 ml MeOH and 25 ml CH2Cl2) and was stirred until completely dissolved. Then hexane was allowed to diffuse slowly into the filtrate at room temperature. Yellow single crystals of 2 could be found in two weeks. A sketched structural relationship is illustrated in Fig. 3.
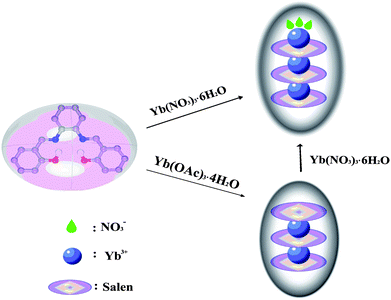 | ||
| Fig. 3 A schematic illustration of the structural relationship between 1 and 2. | ||
NIR photoluminescence experiments on both complexes 1 and 2 in different solvents are conducted (Fig. 4). When excited at 335 nm, the emissions centered at 974 nm and 975 nm are revealed for both complexes 1 and 2, which is attributed to the 2F5/2 → 2F7/2 transition. However, the free salen type ligand does not exhibit any NIR luminescence under the same conditions. Notably, the emission of Yb3+ ion in both 1 and 2 is not a single sharp transition that well-split NIR emission peaks are observed, where it appears as a series of bands with two other broad bands centered at 1003 and 1033 nm. A similar splitting has been reported previously and is attributed to the crystal-field or stark splitting.9 The absence of typical Yb3+ ion excitation bands in the excitation spectra and the ligand-centered luminescence in the emission spectra of 1 and 2 indicate that the ligand-to-metal energy transfer takes place efficiently.
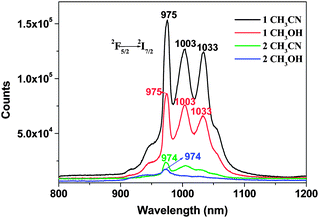 | ||
| Fig. 4 Near infrared luminescence of 1 and 2 in CH3CN (1 × 10−5 M) and CH3OH (1 × 10−5 M) at room temperature. | ||
Obviously, the NIR Yb3+ luminescence intensity of 1 is essentially stronger than that of 2. It may be attributed to the fact that the Yb3+ ions in 1 are completely encapsulated by the salen type ligands which forbid the attachment of the solvent molecules. In 2, three bidentate NO3− anion are considerably exposed, which may partially quench the lanthanide luminescence. In addition, the NIR Yb3+ luminescence intensity of complex 1 in CH3CN is stronger than that in CH3OH. This is attributed to the O–H oscillators in CH3OH, coupling of overtones and the excited state of Yb3+ ion, resulting in the non-radiative transition, which also quench the NIR luminescence.
In addition to the steady-state emission, we also performed time-resolved measurements of complexes 1 and 2 in the NIR region by using the time-correlated single photon counting (TCSPC) technique (Fig. S4 and S5, ESI‡).10 For complex 1, the lifetimes give a satisfactory fit to monoexponential lifetimes with τ1 = 2.339 μs (54.97%) and τ2 = 10.56 μs (45.03%). For complex 2, the corresponding lifetimes are τ1 = 1.566 μs (49.38%) and τ2 = 8.75 μs (50.62%). Obviously, the lifetimes of 1 are longer than that of 2, which is consistent with the intensity of the NIR luminescence. To the best of our knowledge, only two other NIR luminescence lifetime measurements of tetradentate salen type heterodinuclear complexes Zn–Ln (Ln = Nd and Yb) have been previously reported. The first of complex Zn–Yb (at 298 K) is in the range of 11.26–15.89 μs, reported by Loet al. in 2006.11 The second of complex Zn–Nd is in the range of 1.12–1.27 μs, reported by Jiang et al. in 2009.12 Obviously, the lifetime of Yb3+ NIR luminescence in complex Zn–Yb is longer than that of Nd3+ in complex Zn–Nd. The lifetimes of Yb3+ NIR luminescence in complexes 1 and 2 are shorter than that of Yb3+ in 3d–4f heterodinuclear complex Zn–Yb. However, this is the first time that the lifetimes of Yb3+ NIR luminescence in salen type homo-nuclear ytterbium complexes are revealed.
In summary, the isolation of 1 and 2 demonstrates that a tetradentate salen type ligand can stabilize lanthanide ions affording triple-decker dinuclear “real” sandwich lanthanide complexes. The salen moieties of the sandwich structures may work as an antenna, which strengthen the near infrared luminescence of the Yb3+ ion.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 20972043 and 21072049), Heilongjiang Province (No. GC09A402 and 2010td03) and Heilongjiang University.Notes and references
- (a) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, Germany, 1995 Search PubMed; (b) C. Piguet and J.-C. G. Bűnzli, Chem. Soc. Rev., 1999, 28, 347 RSC; (c) A. Døssing, Eur. J. Inorg. Chem., 2005, 1425 CrossRef CAS; (d) K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS; (e) M. Andruh, D. G. Branzea, R. Gheorghe and A. M. Madalan, CrystEngComm, 2009, 11, 2571 RSC.
- (a) S. Natase and J. Reedijk, Coord. Chem. Rev., 2006, 250, 2501 CrossRef CAS; (b) J.-C. G. Bűnzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC; (c) C. Piguet, J.-C. G. Bűnzli, B. Donnio and D. Guillon, Chem. Commun., 2006, 3755 RSC; (d) P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717 CrossRef CAS; (e) X.-P. Yang, R. A. Jones, M. J. Wiester, M. M. Oye and W.-K. Wong, Cryst. Growth Des., 2010, 10, 970 CrossRef CAS; (f) X. Feng, L. Wang, J. Zhao, J. Wang, N. S. Weng, B. Liu and X. Shi, CrystEngComm, 2010, 12, 774 RSC.
- (a) M. P. Hogerheide, J. Boersma and G. van Koten, Coord. Chem. Rev., 1996, 155, 87 CrossRef; (b) A. M. Madalan, H. W. Roesky, M. Andruh, M. Noltemeyer and N. Stanica, Chem. Commun., 2002, 1638 RSC; (c) W. Bi, T. Wei, X. Lü, Y. Hui, J. Song, S. Zhao, W. Wong and R. A. Jones, New J. Chem., 2009, 33, 2326 RSC; (d) A. W. Kleij, Dalton Trans., 2009, 4635 RSC; (e) X.-P. Yang, R. A. Jones, W.-K. Wong, V. Lynch, M. M. Oye and A. L. Holmes, Chem. Commun., 2006, 1836 RSC.
- (a) J.-P. Costes, J.-P. Laussac and F. Nicodème, J. Chem. Soc., Dalton Trans., 2002, 2731 RSC; (b) O. Margeat, P. G. Lacroix, J. P. Costes, B. Donnadieu, C. Lepetit and K. Nakatani, Inorg. Chem., 2004, 43, 4743 CrossRef CAS; (c) J.-P. Costes, A. Dupuis and J.-P. Laurent, Inorg. Chim. Acta, 1998, 268, 125 CrossRef CAS; (d) R. Gheorghe, M. Andruh, J.-P. Costes and B. Donnadieu, Chem. Commun., 2003, 2778 RSC.
- (a) W. Xie, M. J. Heeg and P. G. Wang, Inorg. Chem., 1999, 38, 2541 CrossRef CAS; (b) M. T. Kaczmarek, I. Pospieszna-Markiewicz, M. Kubicki and W. Radecka-Paryzek, Inorg. Chem. Commun., 2004, 7, 1247 CrossRef CAS; (c) W. Radecka-Paryzek, I. Pospieszna-Markiewicz and M. Kubicki, Inorg. Chim. Acta, 2007, 360, 488 CrossRef CAS; (d) X. Yang, R. A. Jones and W.-K. Wong, Chem. Commun., 2008, 3266 RSC; (e) Y.-P. Cai, H.-Z. Ma, B.-S. Kang, C.-Y. Su, W. Zhang, J. Sun and Y.-L. Xiong, J. Organomet. Chem., 2001, 628, 99 CrossRef CAS; (f) X.-P. Yang, B. P. Hahn, R. A. Jones, W. K. Wong and K. J. Stevenson, Inorg. Chem., 2007, 46, 7050 CrossRef CAS; (g) W. Dou, J. Yao, W. Liu, Y. Wang, J. Zheng and D. Wang, Inorg. Chem. Commun., 2007, 10, 105 CrossRef CAS; (h) X.-P. Yang, R. A. Jones, J. H. Rivers and W.-K. Wong, Dalton Trans., 2009, 10505 RSC.
- (a) X.-P. Yang, R. A. Jones, M. M. Oye, A. L. Holmes and W.-K. Wong, Cryst. Growth Des., 2006, 6, 2122 CrossRef CAS; (b) X.-P. Yang and R. A. Jones, J. Am. Chem. Soc., 2005, 127, 7686 CrossRef CAS.
- X.-P. Yang, R. A. Jones and W.-K. Wong, Dalton Trans., 2008, 1676 RSC.
- (a) T. Gao, P.-F. Yan, G.-M. Li, G.-F. Hou and J.-S. Gao, Polyhedron, 2007, 26, 5382 CrossRef CAS; (b) T. Gao, P. Yan, G. Li, G. Hou and J. Gao, Inorg. Chim. Acta, 2008, 361, 2051 CrossRef CAS; (c) W.-B. Sun, P.-F. Yan, G.-M. Li, H. Xu and J.-W. Zhang, J. Solid State Chem., 2009, 182, 381 CrossRef CAS.
- T.-S. Kang, B. S. Harrison, M. Bouguettaya, T. J. Foley, J. M. Boncella, K. S. Schanze and J. R. Reynolds, Adv. Funct. Mater., 2003, 13, 205 CrossRef CAS.
- D. Phillips, R. C. Drake, D. V. O'Connor and R. L. Cristensen, Instrum. Sci. Technol., 1985, 14, 267 CrossRef CAS.
- W.-K. Lo, W.-K. Wong, W.-Y. Wong, J.-P. Guo, K.-T. Yeung, Y.-K. Cheng, X.-P. Yang and R. A. Jones, Inorg. Chem., 2006, 45, 9315 CrossRef CAS.
- H. L. Wang, D. P. Zhang, Z.-H. Ni, X. Y. Li, L. J. Tian and J. Z. Jiang, Inorg. Chem., 2009, 48, 5946 CrossRef CAS.
- B. J. E. Reich, A. K. Justice, B. T. Beckstead, J. H. Reibenspies and Stephen A. Miller, J. Org. Chem., 2004, 69, 1357 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97: Program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Dedicated to Prof. Wenqi Chen on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: UV (Fig. S6), IR (Fig. S7) and excitation spectra (Fig. S8) and decay curves (Fig. S4 and S5). CCDC reference numbers 734804 and 724600 for 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ce00553c |
§ Experimental procedure: the ligand H2L was prepared according to well-established procedures.13 For 1: yellow crystals of 1 suitable for single crystal X-ray diffraction were grown by slowly diffusing the CH3OH solution (15 ml) of Yb(OAc)3·4H2O (0.0422 g, 0.1 mmol) into the CH2Cl2 solution (15 ml) of H2L (0.0633 g, 0.2 mmol). Yield: 0.0665 g (40 wt%). Anal. calcd for 1: C, 55.08; H, 3.56; N, 6.32 wt%. Found: C, 55.47; H, 3.570; N, 6.27 wt%. UV-vis [CH3OH, λmax (nm)]: 241, 289, 385. Conductivity [c = 1 × 10−5 M, 20 C, κ (S m−1)]: 3.0 (CH3OH), 3.7 (CH3CN). For 2: A CH3OH solution (5 ml) of Yb(NO3)3·6H2O (0.0467 g, 0.1 mmol) was added to a CH2Cl2 solution (25 ml) of H2L (0.0633 g, 0.2 mmol). The mixture was stirred at room temperature for 24 h, and the color of the reaction mixture changed from yellow to dark yellow. Hexane or oil ether (60–90 °C) was then allowed to diffuse slowly into the filtrate at room temperature. Yellow single crystals of 2 were harvested in two weeks. Yield 0.0621 g (25 wt%). Anal. calcd for 2: C, 41.25; H, 3.52; N, 6.87 wt%. Found: C, 41.33; H, 3.49; N, 6.95 wt%. UV-vis [CH3OH, λmax (nm)]: 236, 292, 382. Conductivity [c = 1 × 10−5 M, 20 °C, κ (S m−1)]: 7.6 (CH3OH), 8.2 (CH3CN). Single crystals of 1 and 2 of suitable dimensions are mounted onto thin glass fibers. All the intensity data are collected on a Rigaku Raxis-Rapid X-ray diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 293 K. Structures are solved by direct methods followed by difference Fourier syntheses, and then refined by full-matrix least-squares techniques against F2 using SHELXS-97.14 All non-hydrogen atoms are anisotropically refined. Crystal data for 1: C61H47N6O7.5Yb2 (Mr = 1330.13), monoclinic, space groupP21/c, a = 14.504(3) Å, b = 23.595(7) Å, c = 18.676(7) Å, β = 124.20(4)°, V = 5286(3) Å3, Z = 4, Dcalc = 1.671 g cm−3, μ(Mo Kα) = 3.579 mm−1, F(000) = 2619, R1 = 0.0519, wR2 = 0.0998. For 2: C63H64N9O23Yb3 (Mr = 1762.37), triclinic, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 13.785(3) Å, b = 15.175(3) Å, c = 19.161(4) Å, α = 86.08(3)°, β = 71.73(3)°, γ = 73.39(3)°, V = 3646.4(13) Å3, Z = 2, Dcalc = 1.605 g cm−3, μ(Mo Kα) = 3.884 mm−1, F(000) = 1726, R1 = 0.0897, wR2 = 0.1896. CCDC No. 734804 and 724600 contain the supplementary crystallographic data for 1 and 2. , a = 13.785(3) Å, b = 15.175(3) Å, c = 19.161(4) Å, α = 86.08(3)°, β = 71.73(3)°, γ = 73.39(3)°, V = 3646.4(13) Å3, Z = 2, Dcalc = 1.605 g cm−3, μ(Mo Kα) = 3.884 mm−1, F(000) = 1726, R1 = 0.0897, wR2 = 0.1896. CCDC No. 734804 and 724600 contain the supplementary crystallographic data for 1 and 2. |
| This journal is © The Royal Society of Chemistry 2011 |