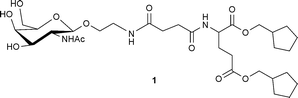
DOI:
10.1039/C0SC00278J
(Edge Article)
Chem. Sci., 2010,
1, 491-498
Supramolecular hydrogel capsule showing prostate specific antigen-responsive function for sensing and targeting prostate cancer cells†
Received
22nd April 2010
, Accepted 26th June 2010
First published on
23rd July 2010
Abstract
A supramolecular hydrogel 1, prepared at high concentration (5–10 wt%), exhibited mechanical toughness comparable to that of a polymer hydrogel, even though the gel was constructed solely by noncovalent bonding interactions among small molecules. The mechanical toughness and thermal reversibility of gel 1 allowed us to fabricate a supramolecular hydrogel capsule (SH-capsule) 1 which was easily handled and was stable in aqueous and cell culture media. The mechanical and substance-release/uptake properties of SH-capsule 1 were investigated by atomic force microscope (AFM) indentation, fluorescence spectroscopy, and confocal laser scanning microscopy (CLSM). On the basis of those properties we successfully designed an enzyme- and cell-responsive SH-capsule. To install a function of enzyme-responsive substance release into the SH-capsule 1, enzyme-labile, amphiphilic additive 2 was embedded in supramolecular nanofibers of 1 through a supramolecular co-assembly method. As a proof-of-concept, we constructed the functional SH-capsule 1/2 that can release a model fluorescent drug triggered by prostate specific antigen (PSA)-catalyzed proteolysis. Selective release of the fluorescent substance was exploited to both assay PSA activity and detect prostate cancer (PCa) cells. We also clearly demonstrated that the released fluorescent substance was delivered and internalized into the PCa cells, mediated by binding to the membrane-bound protein prostate-specific membrane antigen (PSMA), which is over-expressed on a plasma membrane of the PCa cells.
Introduction
Supramolecular hydrogels constructed through the self-assembly of low-molecular-weight (LMW) hydrogelators1 possess several features that are desirable in nanostructured biomaterials. They consist of a three-dimensional (3D) network of well-defined nanofibers. They are easily prepared by a typical self-assembly process, and are smoothly destroyed because of the noncovalent linkage between the LMW building blocks. In addition, biocompatibility is rationally installed in the chemical structure. Furthermore, sophisticated molecular design of LMW-hydrogelators directly reflects the macroscopic properties of the resultant materials. By using these features, supramolecular hydrogels are now actively applied as a unique platform for biosensors,2 matrices for 3D cell culture3 and in controlled drug release.4 Through engineering of LMW-hydrogelators, supramolecular hydrogels have been endowed with sharp and reversible macroscopic gel-to-sol or sol-to-gel transitions in response to a variety of stimuli such as temperature, pH,5 chemicals,6 metal cations,7 anions8 and mechanical force,3g light9 and enzymes.10,11 Most supramolecular hydrogels, however, do not have mechanical toughness comparable to that of polymer-based hydrogels, and their lack of mechanical toughness unfortunately limits their potential utility as novel biomaterials.12
Among many stimuli-responsive functions, enzyme-response is of great interest for biomaterials applications because many disease-related enzymes have been now unveiled.13,14 Xu and others pioneered an enzyme-triggered sol-to-gel transition10,11 and van Esch et al.11a reported enzyme-regulated drug release from a supramolecular hydrogel. However, compared with a representative example of polymer-based hydrogels15 reported by Hubbell and co-workers,15a who elegantly accomplished repair of bone defects by enzyme- and cell-responsive drug release systems, enzyme-responsive materials based on supramolecular hydrogels have not yet been explored to a significant extent. Considering recent progress in the fundamentals of supramolecular hydrogels, more sophisticated biomaterials applicable to diagnosis or medical treatments are keenly anticipated.
We report herein a supramolecular hydrogel comprising glycolipid mimic 1, prepared at high concentration, that exhibits sufficient stability and mechanical toughness (comparable to that of agarose-polymer gel) for various shapes, including a hydrogel capsule, to be easily fabricated and used in water and cell culture media. Using a supramolecular co-assembly method,16 by means of which we previously equipped glycolipid-based hydrogels with pH responsiveness5c and enhanced mechanical toughness,17 an enzyme-labile additive was embedded in the hydrogel fibers of the capsule. We thus successfully developed a hydrogel capsule responsive to prostate specific antigen (PSA),14 a representative biomarker enzyme secreted from prostate cancer (PCa) cells, by co-assembly of a PSA-cleavable additive with hydrogel matrix 1. The functionalized capsule displayed the PSA-responsive release of a fluorescent substance, which allowed us not only to assay the PSA activity, but also to selectively detect PCa cells. Furthermore, we demonstrated that the released substance was delivered and internalized into PCa cells with the help of the tethered ligand targeting a membrane-bound protein over-expressed on the PCa cell surface. The present study clearly demonstrates that elaborate modification of stiff and stable supramolecular hydrogel capsules is a promising route to novel biofunctional materials.
Results and discussion
Mechanical properties of a supramolecular hydrogel that can be prepared at high concentration
Polymer hydrogels with substantial mechanical toughness are usually prepared at about 10 wt% concentration,15a,18 whereas a supramolecular hydrogel is generally prepared at lower concentration.1 While simple increase of the hydrogelator concentration might be expected to give a mechanically tough supramolecular hydrogel, most supramolecular hydrogels at high concentration precipitate and/or crystallize. In the present study, we re-screened a small library of glycolipid mimics previously reported by us,2a,3h,9c,19 and found that hydrogelator 1 can form a stable hydrogel even at 10 wt%. Other hydrogelators could not be dissolved at such a high concentration3h,9c or resulted in crystals.2a Hydrogelator 1 can be dissolved completely at more than 5 wt% concentration with gentle heating, and hydrogel 1 formed after cooling for several minutes exhibited a typical viscoelastic feature of hydrogels (Fig. 1A), and sufficient mechanical toughness for it to be easily handled (inset of Fig. 1A). The presence of a 3D network of nanofibers bearing continuous hydrophobic domains inside the hydrogel was confirmed by confocal laser scanning microscopy (CLSM) observation.2a,19 From Fig. 1B, it is clear that the storage modulus G′ and the yield stress (σc) of hydrogel 1 dramatically increased from 0.24 to 63 kPa (260-fold) and 17.8 to 631 Pa (35-fold), respectively, by increasing the concentration of 1 from 0.5 to 5.0 wt%. The G′ value (0.63 MPa) is remarkably high for supramolecular hydrogels reported thus far1,20 and the σc value is comparable with that of agarose polymer gel.21
![Oscillatory rheology for (A) frequency sweeps and (B) amplitude-sweep of supramolecular hydrogel 1 (A: [1] = 5.0 wt%, B: [1] = 0.5 (blue) and 5.0 wt% (red), in ion-exchanged water) at 25 °C. The inset in panel A shows a fabricated supramolecular hydrogel ([1] = 5.0 wt%, in ion-exchanged water).](/image/article/2010/SC/c0sc00278j/c0sc00278j-f1.gif) |
| | Fig. 1 Oscillatory rheology for (A) frequency sweeps and (B) amplitude-sweep of supramolecular hydrogel 1 (A: [1] = 5.0 wt%, B: [1] = 0.5 (blue) and 5.0 wt% (red), in ion-exchanged water) at 25 °C. The inset in panel A shows a fabricated supramolecular hydrogel ([1] = 5.0 wt%, in ion-exchanged water). | |
Because of its mechanical toughness, a spherically-shaped supramolecular hydrogel, referred to hereafter as a supramolecular hydrogel capsule (SH-capsule), was easily prepared by injecting a hot aqueous sol (several μL) of 1 (10 wt%, Tgel = 52 °C) into an oil, hexadecane (HD), at room temperature according to the method reported previously (Fig. 2A).9d,22 The SH-capsules 1 obtained were easily handled (Fig. 2B) and stable in water without dissolution.23 The spherical shape of the SH-capsule 1 was clearly confirmed by CLSM images (Fig. 2B bottom). By contrast, capsules prepared at concentrations lower than 1.0 wt% were fragile and quickly dispersed to be eventually dissolved in water, which indicates that densely entangled nanofibers inside the hydrogel at high concentration are indispensable for SH-capsules to be stable in water. We thus employed the SH-capsule prepared at 10 wt% in subsequent experiments. In addition to its enhanced stability in water, we also examined the mechanical properties of the SH-capsule 1 using atomic force microscope (AFM) indentation experiments.24Fig. 2C shows typical force indentation curves for gel capsules, yielding the average Young's modulus values (E, mean ± s.d.) 4.4 ± 1.8 and 8.1 ± 3.4 kPa for the SH-capsule 1 and an agarose gel capsule, respectively (Fig. 2D). These values are in the range of the moduli for bulk agarose gels previously evaluated by AFM indentation.25 It is concluded that a SH-capsule 1 fabricated at the several hundred micrometre size has a mechanical toughness comparable to that of an agarose gel capsule of similar size.
![(A) Scheme for preparation of SH-capsule 1 by injecting a heated sol of 1 into HD. (B) Photograph of SH-capsules 1 (1: 0.2 μL, 2: 0.5 μL, 3: 1.0 μL, 4: 2.0 μL, [1] = 10 wt%, [BODIPY] = 50 μM (see ESI for the molecular structure), scale bar = 1.0 mm) and CLSM images (fluorescence and DIC mode) of SH-capsule 1 (2.0 μL). The fluorescent dye (BODIPY) was encapsulated to clearly visualize SH-capsule 1; scale bar = 500 μm. (C) Typical force indentation curves of hydrogel capsules (SH-capsule 1 (10 wt%) and agarose gel capsule (1.0 wt%)). The solid lines represent the fit by a conical tip model. (D) Averaged Young's modulus E (kPa) for all data collected from three different samples of SH-capsule 1 and agarose gel capsule (n = 33 for SH-capsule 1; n = 30 for agarose gel capsule).](/image/article/2010/SC/c0sc00278j/c0sc00278j-f2.gif) |
| | Fig. 2 (A) Scheme for preparation of SH-capsule 1 by injecting a heated sol of 1 into HD. (B) Photograph of SH-capsules 1 (1: 0.2 μL, 2: 0.5 μL, 3: 1.0 μL, 4: 2.0 μL, [1] = 10 wt%, [BODIPY] = 50 μM (see ESI† for the molecular structure), scale bar = 1.0 mm) and CLSM images (fluorescence and DIC mode) of SH-capsule 1 (2.0 μL). The fluorescent dye (BODIPY) was encapsulated to clearly visualize SH-capsule 1; scale bar = 500 μm. (C) Typical force indentation curves of hydrogel capsules (SH-capsule 1 (10 wt%) and agarose gel capsule (1.0 wt%)). The solid lines represent the fit by a conical tip model. (D) Averaged Young's modulus E (kPa) for all data collected from three different samples of SH-capsule 1 and agarose gel capsule (n = 33 for SH-capsule 1; n = 30 for agarose gel capsule). | |
Substance entrapment and penetration properties of SH-capsule
To evaluate its potential utility as a drug release reservoir, the entrapment capacity of the SH-capsule 1 was next studied using small molecular dyes and proteins of varying molecular weight (Fig. 3A). Fig. 3B shows the release profiles of entrapped substances, as graphs of the fraction released (Mt/M∞) as a function of time. The results, including kinetic parameters obtained by use of the Higuchi model, are summarized in Table 1.26 Among the small molecules, hydrophilic dyes such as fluorescein and rhodamine 110 were released rapidly and quantitatively, indicating that hydrophilic dyes can move without significant restriction inside the supramolecular hydrogel 1. By contrast, release of the hydrophobic dye pyrene butyric acid was significantly suppressed, which may be due to trapping of the hydrophobic pyrene moiety within supramolecular fibers through hydrophobic interactions.28 Such an encapsulation ability, depending on the hydrophobicity of the entrapped guest molecule, seems to be one of the important features of this SH-capsule, bearing a hydrophobic nanofiber interior.29 The speed of release of water-soluble proteins was roughly dependent on their molecular weight, that is, the smaller the protein the faster its release. Protein release did not reach 100% (especially for immunoglobulin G (IgG)), which may be due to some fractions of the protein being physically entrapped in highly entangled nanofiber networks or hydrophobic domains of the protein being adsorbed on hydrophobic domains of the nanofibers. Overall, the SH-capsule exhibited a unique encapsulation ability for substances depending on their molecular weight and hydrophobicity.
![(A) Chemical structures of dyes used in release experiments. (B) Release profiles for dyes and proteins from the SH-capsule 1 in PBS (phosphate buffered saline) at 25 °C. [1] = 10 wt%, 2.0 μL, [substance] = 5.0 μM for initial concentration in SH-capsule 1.](/image/article/2010/SC/c0sc00278j/c0sc00278j-f3.gif) |
| | Fig. 3 (A) Chemical structures of dyes used in release experiments. (B) Release profiles for dyes and proteins from the SH-capsule 1 in PBS (phosphate buffered saline) at 25 °C. [1] = 10 wt%, 2.0 μL, [substance] = 5.0 μM for initial concentration in SH-capsule 1. | |
Table 1 Properties of dyes and proteins encapsulated in SH-capsule 1 and diffusion kinetic parameters estimated from release profiles
| Substance |
M
w/Da |
Net chargea or pIb |
Release (%) at 60 min |
k (10−2 s−1/2) |
| Fluorescein |
323 |
−2a |
97 ± 3 |
2.7 |
| Rhodamine 110 |
367 |
0a |
95 ± 5 |
1.8 |
| Pyrene butyric acid |
288 |
−1a |
33 ± 1 |
0.43 |
| FITC-trypsin |
23.3 k |
10.1b |
75 ± 1 |
1.6 |
| FITC-Con A |
100 k |
8.4b |
70 ± 5 |
1.6 |
| FITC-IgG |
ca. 150 k |
ca. 7.2b |
18 ± 3 |
0.45 |
In addition to substance entrapment, the permeability from bulk solution to the capsule interior is another important factor in the design of functional SH-capsules. Thus we evaluated the ability of proteins to penetrate the SH-capsule 1. The diffusion of the proteins FITC-labeled trypsin, concanavalin A (Con A), and IgG from the bulk aqueous phase into the capsule was monitored using CLSM observations. The CLSM images clearly showed that all FITC-labeled proteins diffused into the SH-capsule, and the smaller the protein the larger the rate of diffusion (ESI†, Fig. S3). It is clear that the molecular weight cut-off (MWCO) of the SH-capsule 1 for proteins was above 100 kDa, which is higher than that of poly(ethylene glycol) (PEG) hydrogel-based capsules (<40 kDa).18 This implies that larger enzymes can penetrate into the SH-capsule, compared with the polymer gel capsule.
Design of SH-capsule that can respond to prostate specific antigen (PSA) by co-assembly
On the basis of the aforementioned fundamental properties of the SH-capsule 1, we developed a design strategy (depicted in Fig. 4) for constructing functional SH-capsules capable of sensing and targeting PCa. PCa is the most frequently diagnosed cancer for men (approximately 2000 men per day worldwide).30 Thus, rapid and convenient diagnostic tools and drug release materials for PCa are highly desirable. Since PSA (33 kDa), a specific protease secreted from PCa, is smaller than the MWCO of the SH-capsule 1, it is conceivable that PSA can smoothly diffuse into the interior of the SH-capsule. A PSA-labile additive 2 was designed, which contains PCa-cell targeting DUPA (2-[3-(1,3-dicarboxypropyl)ureido]pentanedioic acid)31 and a pyrene moiety linked through a PSA specific peptide substrate. The pyrene moiety of 2 was expected to act as a binding anchor to supramolecular nanofibers, since pyrene butyric acid was entrapped sufficiently in the SH-capsule 1 (vide supra). Upon penetration of PSA into the SH-capsule 1, the proteolytic cleavage of 2 tethered to supramolecular nanofibers can release the hydrophilic fragment 3 from the SH-capsule 1 to aqueous solution (scheme enclosed in rectangle in Fig. 4). As the PSA specific peptide linker, a sequence SSIY↓SQTEEQ (kcat/Km = 61 mM−1 s−1) was used as previously reported.33 TAMRA (tetramethylrhodamine), a fluorescent marker, was tethered as a drug model to trace the localization and amount of 2 and 3 by both CLSM observation and fluorescence spectroscopy measurements. Monitoring the released TAMRA fluorescence in the supernatant allowed us to sense the PSA activity and consequently the presence or absence of PCa. For targeting, we focused on PSMA, a plasma membrane-associated glycoprotein (Mr ∼100 kDa), which is selectively over-expressed on the cell surface of the vast majority of PCa-cells.34 Since PSMA is known to undergo internalization through clathrin-coated pits, it is regarded as a suitable candidate for PCa-targeting drug delivery.35 For that reason we also attached DUPA, a PSMA-targeting ligand, to the N-terminal of 2 as a PCa cell-targeting unit.32 It was reasonably expected that released 3 could bind selectively to PSMA on the membrane surface of a PSMA-positive PCa cell such as LNCaP, and subsequently internalize into the cell owing to DUPA. This strategy could enhance the selectivity of the released substance toward a cancer cell without affecting healthy cells.
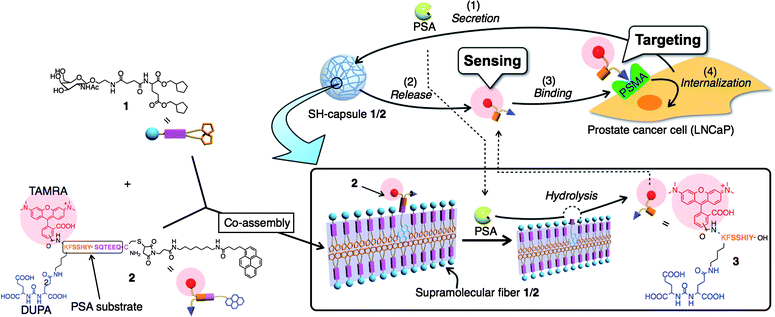 |
| | Fig. 4 Schematic illustration of SH-capsule 1/2 for sensing and targeting PCa cells. Supramolecular nanofibers 1/2 can release a PSMA binding substance (3) in response to hydrolytic cleavage by PSA secreted from a prostate cancer cell (LNCaP). The released substance 3 can bind selectively to PSMA on the cell surface of LNCaP, and subsequently internalize into the cell. | |
Sensing PSA enzyme and LNCaP cells using enzyme-responsive release of fluorescent TAMRA from SH-capsule 1/2
For the preparation of the SH-capsule 1 functionalized with 2, we applied the co-assembly method. Mixing a hot aqueous sol of 1 with a dimethyl sulfoxide (DMSO) solution of 2, followed by injection of the solution into HD, gave rise to SH-capsule 1/2 (mol% of 2 against 1 is 0.033%, 2 μL, 1696 ± 23 μm in diameter). The additive 2 was embedded in the nanofibers 1 through hydrophobic interactions as for pyrene butyric acid and was thus trapped inside the capsule almost quantitatively (less than 5% leakage over 360 min; ◆ in Fig. 5A, D) against buffers or cell culture medium such as RPMI 1640 (RPMI = Roswell Park Memorial Institute) supplemented with 10% fetal bovine serum (FBS) at room temperature.36 The AFM indentation study and the stability in water indicated that the influence of DMSO and co-assembly of 2 on the mechanical properties and its stability was negligible under these conditions (ESI†, Fig. S2, S4). When capsule 1/2 was dropped into an aqueous solution containing PSA ([PSA] = 500 nM), the fluorescence intensity of the supernatant increased during a 360 min period (red closed circles in Fig. 5A).37 The release rate was clearly dependent on the PSA concentration (Fig. 5B). With addition of trypsin instead of PSA, however, a substantial increase in fluorescence intensity was not observed. Similarly, in the presence of an inhibitor for PSA (IC50 = 0.5 μM, see ESI† for the molecular structure),38 the fluorescence intensity increase was suppressed. CLSM observations inside the SH-capsule 1/2 (Fig. 6A) clearly showed fibrous morphologies arising from TAMRA fluorescence before PSA addition, whereas the most fibrous morphologies were smeared after PSA treatment for 6 h, in good agreement with the release profile of 3 shown in Fig. 6A,B. These lines of evidence strongly support the scheme shown in Fig. 4, that is, selective cleavage of 2 embedded with the supramolecular nanofiber 1 through hydrophobic interactions was catalyzed by PSA that penetrated the SH-capsule 1/2 and this proteolysis produced a hydrophilic fragment 3 that was released from the capsule. Thus it was possible to sense the PSA activity by monitoring the increase in the TAMRA fluorescence intensity of the supernatant. A photograph of PSA-selective release of the fluorescent 3 from the SH-capsule 1/2 clearly revealed the presence of PSA in solution by naked eye, that is, strong fluorescence was observed in the supernatant including PSA (Fig. 5C(b)), whereas it remained in the SH-capsule 1/2 in the solution without PSA (Fig. 5C(a)). Fig. 5B suggests that the detection limit of PSA is roughly between 5 and 50 nM. Although an ELISA-based assay is more sensitive,14b, 39 the present system is remarkably simple and thus is applicable under various conditions and can evaluate the amount of catalytically active PSA.
![(A, D) Release profiles traced by TAMRA fluorescence from SH-capsule 1/2 ([1] = 10 wt% (152 mM), [2] = 50 μM in 2.0 μL. A: 50 mM Tris-HCl (pH 8.3), 25 mM NaCl, [PSA inhibitor] = 10 μM. D: RPMI 1640 with 10% FBS, 25 mM HEPES (pH 7.4 for LNCaP and Jurkat and DMEM with 10% FBS, 25 mM HEPES (pH 7.4) for HeLa, Hep G2, A549, and HEK 293) at room temperature (ESI, Fig. S7). (B, E) Release (%) at 360 min for different conditions corresponding to A and D (*P = 0.0004 using Student's t-test, n = 3). (C) Photograph (under light at 365 nm) of aqueous solutions (a: no PSA, b: [PSA] = 500 nM) incubated with SH-capsule 1/2 at the bottom of quartz cells for 12 h at room temperature.](/image/article/2010/SC/c0sc00278j/c0sc00278j-f5.gif) |
| | Fig. 5 (A, D) Release profiles traced by TAMRA fluorescence from SH-capsule 1/2 ([1] = 10 wt% (152 mM), [2] = 50 μM in 2.0 μL. A: 50 mM Tris-HCl (pH 8.3), 25 mM NaCl, [PSA inhibitor] = 10 μM. D: RPMI 1640 with 10% FBS, 25 mM HEPES (pH 7.4 for LNCaP and Jurkat and DMEM with 10% FBS, 25 mM HEPES (pH 7.4) for HeLa, Hep G2, A549, and HEK 293) at room temperature (ESI†, Fig. S7). (B, E) Release (%) at 360 min for different conditions corresponding to A and D (*P = 0.0004 using Student's t-test, n = 3). (C) Photograph (under light at 365 nm) of aqueous solutions (a: no PSA, b: [PSA] = 500 nM) incubated with SH-capsule 1/2 at the bottom of quartz cells for 12 h at room temperature. | |
![CLSM images (fluorescence mode: TAMRA) of supramolecular nanofiber 1/2 ([1] = 10 wt%, [2] = 50 μM in 2.0 μL) (A) before, and (B) after treatment with PSA ([PSA] = 500 nM, after 6 h, room temperature). The image (B) was obtained with higher laser intensity because the fluorescence intensity inside the SH-capsule 1/2 was substantially decreased (ESI, Fig. S8).](/image/article/2010/SC/c0sc00278j/c0sc00278j-f6.gif) |
| | Fig. 6 CLSM images (fluorescence mode: TAMRA) of supramolecular nanofiber 1/2 ([1] = 10 wt%, [2] = 50 μM in 2.0 μL) (A) before, and (B) after treatment with PSA ([PSA] = 500 nM, after 6 h, room temperature). The image (B) was obtained with higher laser intensity because the fluorescence intensity inside the SH-capsule 1/2 was substantially decreased (ESI†, Fig. S8). | |
An increase in fluorescence intensity of the supernatant was also observed when the SH-capsule 1/2 was dropped into a cell culture medium after cultivating LNCaP cells, suggesting that the secreted PSA from LNCaP behaved in a manner similar to purified PSA (Fig. 5D). By contrast, cell culture media that cultivated HeLa, Jurkat, HEK 293, A 549, and Hep G2 cells for 4 days, all of which do not secrete PSA, did not induce an increase in fluorescence intensity, indicating that the release of 3 from the SH-capsule 1/2 is highly selective toward LNCaP cells (Fig. 5E). These results implied that the SH-capsule 1/2 can be utilized as a sensing material not only for PSA enzyme but also for LNCaP cells. The stability of SH-capsule 1/2 and PSA-selective cleavage even in crude aqueous media40 are critically attributed to these selective release and sensing functions.
PSA-responsive targeting of released substances from SH-capsule 1/2 to a prostate cancer (PCa) cell
The live cell targeting of 3 released from SH-capsule 1/2 was found by placing the capsules 1/2 directly in a culture dish containing PSA-secreting and PSMA-positive LNCaP cells (ESI†, Fig. S9). There was no apparent cytotoxicity of the SH-capsule 1/2 (data not shown). The fluorescence intensity coming from TAMRA of the fragment 3 around LNCaP cells increased constantly, confirming that PSA secreted from LNCaP cells induced the release of 3 from the capsule (Fig. 7A, see also ESI†, S10). More importantly, strong fluorescence was observed after 6 h in the cytoplasm of LNCaP cells as well as on the cell surface, in CLSM images (Fig. 7B). In the presence of an excess amount of the competing ligand (PMPA,42,43Ki = 0.3 nM, see ESI† for the molecular structure) for PSMA,44 by contrast, no fluorescence internally and on the surface of LNCaP cells was observed before and after the medium exchange (Fig. 7B). These results indicate that the released 3 was delivered to and transported into the LNCaP cells through selective binding of DUPA to PSMA on the cell membrane, followed by internalization.43
![(A) Change in fluorescence intensity around LNCaP and HeLa cells as a function of time evaluated by time lapse CLSM images (ESI, Fig. S9A, 10A) of LNCaP and HeLa cells incubated with SH-capsules 1/2 ([1] = 10 wt%, [2] = 50 μM in 2.0 μL). (B) Magnified CLSM images of LNCaP cells after 6 h incubation with SH-capsules 1/2 in the absence or presence of PMPA at room temperature followed by medium exchanges ([PMPA] = 50 μM).](/image/article/2010/SC/c0sc00278j/c0sc00278j-f7.gif) |
| | Fig. 7 (A) Change in fluorescence intensity around LNCaP and HeLa cells as a function of time evaluated by time lapse CLSM images (ESI†, Fig. S9A, 10A) of LNCaP and HeLa cells incubated with SH-capsules 1/2 ([1] = 10 wt%, [2] = 50 μM in 2.0 μL). (B) Magnified CLSM images of LNCaP cells after 6 h incubation with SH-capsules 1/2 in the absence or presence of PMPA at room temperature followed by medium exchanges ([PMPA] = 50 μM). | |
As control experiments, when the capsules 1/2 were placed in a dish containing HeLa cells, which neither secrete PSA nor express PSMA, the fluorescence intensity around the HeLa cells did not change (ESI†, Fig. S11A,B). In addition, when fragment 3 was incubated with HeLa cells, TAMRA fluorescence was detected neither inside nor at the membrane of HeLa cells after the medium exchange (ESI†, Fig. S11C), indicating that 3 is not capable of binding to and entering live HeLa cells. These results clearly demonstrate that release and delivery of a substance selective to the PSMA-positive PCa cell was carried out by SH-capsule 1/2.
Conclusions
We succeeded in constructing an SH-capsule 1/2 that is capable of sensing and targeting PCa cells. It is clear that the enhanced mechanical toughness of supramolecular hydrogels by increasing the gelator concentration is crucial to their applications as biomaterials. This study also demonstrated that a supramolecular self-assembly and co-assembly approach, powerful for fabricating and flexibly tuning nanostructured functional biomaterials, requires careful and suitable molecular design of an additive for a desired function. Controlling the amphiphilic character of the additive is critical for optimizing the location and/or the orientation of the additive embedded in the supramolecular nanofibers 1. Taking advantage of the present modular strategy for the SH-capsule construction, we envisage that installing other stimuli-responsiveness5–9 and/or cell-targeting properties in SH-capsules may further expand the general utility of the unique semi-wet soft materials for applications such as intelligent controlled release and cell culture matrices and actuators. We are pursuing this line of research.
Experimental procedures
Unless stated otherwise, all commercial reagents were used as received. 5- (or 6)-carboxy-tetramethylrhodamine-succimidyl ester (5/6-TAMRA SE) and PSA were purchased from Invitrogen and Calbiochem, respectively. Thin layer chromatography (TLC) was performed on silica gel 60F254 (Merck). Column chromatography was performed on silica gel 60N (Kanto, 40–50 μm). Reverse phase HPLC (RP-HPLC) was conducted with a Hitachi Lachrom instrument equipped with YMC-pack ODS-A columns (250 mm × 20 mm I.D. or 250 mm × 4.6 mm I. D.). 1H and 13C NMR spectra were obtained on a Varian Mercury 400 spectrometer with tetramethylsilane (TMS) or residual non-deuterated solvents as the internal references. FAB, MALDI-TOF, and ESI mass spectra were recorded using a Shimadzu QP5050A (NBA as a matrix), Bruker autoflexIII (CHCA or DHB as a matrix), and Applied Biosystems API2000, respectively. The absorption and fluorescence spectra were measured using a Shimadzu UV2550 and a Perkin-Elmer LS55 spectrometer, respectively. Rheological measurements were carried out using a Reologica DynAlyser DAR-100 with a parallel plate (diameter = 4.0 cm) at strain (0.10%) and gap (1.6 mm) at 24 °C and the reproducibility of the data was confirmed by using at least two individual samples. Elemental analysis was carried out by the services at Kyoto University.
Synthesis of 2
A solution of maleimide-appended tail 5 (7.0 mg, 13 μmol) and TAMRA-K(DUPA)FSSIYSQTEEQC-CONH2 peptide (20 mg) in DMSO/PBS buffer (pH 7.2) (1 mL/100 μL) was incubated at room temperature (Scheme 1). After 2 days, the mixture was purified by RP-HPLC (wavelength of detection: 220 nm, eluent: A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B = 70:30 to 100:0 (A: acetonitrile/0.1% TFA, B: H2O/0.1% TFA), linear gradient over 40 min, flow rate = 9.9 mL min−1, tR = 16.1 min). Subsequent lyophilization afforded compound 2 as a purple solid (4.5 mg, 21%). MALDI-MS (DHB): calcd. for [M]+: m/z = 2814.2; found: 2815.0 (see ESI†, Fig. S1 for analytical RP-HPLC of 2).
:B = 70:30 to 100:0 (A: acetonitrile/0.1% TFA, B: H2O/0.1% TFA), linear gradient over 40 min, flow rate = 9.9 mL min−1, tR = 16.1 min). Subsequent lyophilization afforded compound 2 as a purple solid (4.5 mg, 21%). MALDI-MS (DHB): calcd. for [M]+: m/z = 2814.2; found: 2815.0 (see ESI†, Fig. S1 for analytical RP-HPLC of 2).
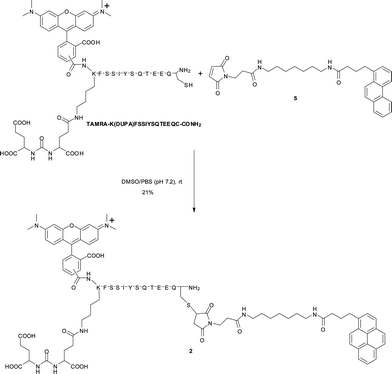 |
| | Scheme 1 Synthesis of 2. | |
Evaluating mechanical properties of gel capsules using AFM
The mechanical properties of gel capsules in water at ambient temperature were characterized by indentation measurements using an AFM microscope (Shimadzu SPM-9600) equipped with a silicon nitride probe (TR, Olympus, a spring constant of 0.15 N m−1) in force mode. Force–displacement curves were recorded at 1 Hz. Young's moduli (E (Pa)) were calculated by fitting force–indentation curves converted from the force curves with the Hertz model, which was based on the indentation of an elastic sample using a stiff conical indenter.24,25 The half opening angle of the AFM tip was 35° and the Poisson ratio was taken to be 0.5, as is typical for soft materials.
Substance release and penetration of SH-capsule 1
For substance release experiments, an aqueous solution of 1 (11 wt%, 18 μL) was mixed with an aqueous solution of fluorescent substance ([substance] = 50 μM, 2.0 μL) and the resulting solution (2.0 μL) was dropped into HD to form SH-capsule ([1] = 10 wt%, [substance] = 5.0 μM) and incubated to complete gelation at room temperature for 2 h. The SH-capsule 1 (10 wt%, 2.0 μL) containing a fluorescent substance (5.0 μM) was directly transferred into a quartz cell. PBS (498 μL) was carefully added into the quartz cell and fluorescence released into supernatant was traced by fluorescence spectrometer. The concentration of the fluorescent substance released from the SH-capsule was determined using a calibration curve of the pure substance in PBS under the same conditions. For substance uptake experiments, SH-capsule 1 (10 wt%, 2.0 μL) was transferred into a glass-bottomed dish (Matsunami). An aqueous solution of FITC-labeled protein (2.0 μM, 50 μL) was carefully added to the dish and the sample was subject to CLSM observations. An inverted CLSM (Olympus FV1000-ASW) with a 100×, NA = 1.40 oil objective or a 4×, NA = 0.16 air objective and 543 nm and 633 nm Helium Neon or 488 nm Ar laser was employed to obtain images.
PSA and LNCaP sensing with SH-capsule 1/2
SH-capsule 1/2 ([1] = 10 wt%, 2.0 μL, [2] = 50 μM) was directly transferred into a quartz cell. An aqueous solution of PSA or a cell-culture medium (398 μL) which was collected from a dish used to cultivate cells (initial condition; 5 × 105 cells/well in 1 mL) for 4 days, was carefully added into the quartz cell and fluorescence released into the supernatant was traced by fluorescence spectrometer. The concentration of the fluorescent substance 3 released from the SH-capsule was determined using a calibration curve of 3 obtained by PSA treatment without the SH-capsule under the same conditions.
Live cell imaging
LNCaP cells or HeLa cells (5 × 105 cells/well in 1 mL) were seeded into a poly-L-lysine treated glass-bottomed dish (IWAKI) and cells allowed to form monolayers over 4 days. Then, four SH-capsules 1/2 ([1] = 10 wt%, 2.0 μL, [2] = 50 μM) were transferred into the dish and the sample was subjected to CLSM observation (ESI†, Fig. S9).
Acknowledgements
We thank Dr T. Sakamoto (JAIST) for his helpful comments and discussions. We thank Prof. T. Takigawa and Prof. K. Urayama (Kyoto University) for rheological measurements and the Shimadzu Corporation for AFM indentation experiments. We gratefully acknowledge financial support from the JST (Japan Science and Technology Agency), CREST program and the global COE program, “Integrated Materials Science” of the Ministry of Education, Culture, Science, Sports, and Technology (Japan).
Notes and references
- For reviews on supramolecular hydrogels, see:
(a) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS
 ;
(b) M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615 CrossRef CAS ;
(c) Z. Yang and B. Xu, J. Mater. Chem., 2007, 17, 2385 RSC ;
(d) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS and references cited therein.
;
(b) M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615 CrossRef CAS ;
(c) Z. Yang and B. Xu, J. Mater. Chem., 2007, 17, 2385 RSC ;
(d) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS and references cited therein.
-
(a) S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58 CrossRef CAS ;
(b) I. Yoshimura, Y. Miyahara, N. Kasagi, H. Yamane, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 12204 CrossRef CAS ;
(c) Z. Yang and B. Xu, Chem. Commun., 2004, 2424 RSC ;
(d) S. Yamaguchi, I. Yoshimura, T. Kohira, S.-i. Tamaru and I. Hamachi, J. Am. Chem. Soc., 2005, 127, 11835 CrossRef CAS ;
(e) S.-i. Tamaru, S. Kiyonaka and I. Hamachi, Chem.–Eur. J., 2005, 11, 7294 CrossRef CAS ;
(f) Y. Koshi, E. Nakata, H. Yamane and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 10413 CrossRef CAS ;
(g) A. Wada, S.-i. Tamaru, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5321 CrossRef CAS ;
(h) M. Ikeda, R. Ochi and I. Hamachi, Lab Chip, 2010 10.1039/c004908e .
-
(a) S. Zhang, T. C. Holmes, C. M. DiPersio, R. O. Hynes, X. Su and A. Rich, Biomaterials, 1995, 16, 1385 CrossRef CAS ;
(b) T. C. Holmes, S. de Lacalle, X. Su, G. Liu, A. Rich and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728 CrossRef CAS ;
(c) G. A. Silva, C. Czeisler, K. L. Niece, E. Beniach, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352 CrossRef CAS ;
(d) V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611 CrossRef CAS ;
(e) A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365 CrossRef CAS ;
(f) F. Gelain, A. Horii and S. Zhang, Macromol. Biosci., 2007, 7, 544 CrossRef CAS and references cited therein;
(g) L. Haines-Butterick, K. Rajogopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791 CrossRef CAS ;
(h) M. Ikeda, S. Ueno, S. Matsumoto, Y. Shimizu, H. Komatsu, K.-i. Kusumoto and I. Hamachi, Chem.–Eur. J., 2008, 14, 10808 CrossRef CAS .
-
(a) H. Komatsu, S. Matsumoto, S.-i. Tamaru, K. Kaneko, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5580 CrossRef CAS ;
(b) G. Liang, Z. Yang, R. Zhang, L. Li, Y. Fan, Y. Kuang, Y. Gao, T. Wang, W. W. Lu and B. Xu, Langmuir, 2009, 25, 8419 CrossRef CAS ;
(c) S. Koutsopoulos, L. D. Unswortha, Y. Nagai and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4623 CrossRef CAS ;
(d) J. Naskar, G. Palui and A. Banerjee, J. Phys. Chem. B, 2009, 113, 11787 CrossRef CAS .
-
(a) A. Aggeli, M. Bell, L. M. Carrick, C. W. G. Fishwick, R. Harding, P. J. Mawer, S. E. Radford, A. E. Strong and N. Boden, J. Am. Chem. Soc., 2003, 125, 9619 CrossRef CAS ;
(b) K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663 CrossRef CAS ;
(c) S.-L. Zhou, S. Matsumoto, H.-D. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, Chem.–Eur. J., 2005, 11, 1130 CrossRef CAS .
-
(a) W. Deng, H. Yamaguchi, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2007, 46, 5144 CrossRef CAS ;
(b) M. Masuda, K. Sumitomo, H. Minamikawa, N. Kameta, Y. Yamaguchi and T. Shimizu, Chem. Lett., 2009, 38, 606 CrossRef CAS .
-
(a) N. Sreenivasachary and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938 CrossRef CAS ;
(b) W. Weng, Z. Li, A. M. Jamieson and A. J. Rowan, Macromolecules, 2009, 42, 236 CrossRef CAS ;
(c) I. Imaz, M. Marta Rubio-Martínez, W. J. Saletra, D. B. Amabilino and D. Maspoch, J. Am. Chem. Soc., 2009, 131, 18222 CrossRef CAS .
-
(a) T. Becker, C. Y. Goh, F. Jones, M. J. McIldowie, M. Mocerino and M. I. Ogden, Chem. Commun., 2008, 3900 RSC ;
(b) H.-J. Kim, J.-H. Lee and M. Lee, Angew. Chem., Int. Ed., 2005, 44, 5810 CrossRef CAS .
-
(a) L. Frkanec, M. Jokić, J. Makarević, K. Wolsperger and M. Žinić, J. Am. Chem. Soc., 2002, 124, 9716 CrossRef CAS ;
(b) L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025 CrossRef CAS ;
(c) S. Matsumoto, S. Yamaguchi, S. Ueno, H. Komatsu, M. Ikeda, K. Ishizuka, Y. Iko, K. V. Tabata, H. Aoki, S. Ito, H. Noji and I. Hamachi, Chem.–Eur. J., 2008, 14, 3977 CrossRef CAS ;
(d) S. Matsumoto, S. Yamaguchi, A. Wada, T. Matsui, M. Ikeda and I. Hamachi, Chem. Commun., 2008, 1545 RSC ;
(e) Z. Qiu, H. Yu, J. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342 RSC ;
(f) T. Muraoka, C.-Y. Koh, H. Cui and S. I. Stupp, Angew. Chem., Int. Ed., 2009, 48, 5946 CrossRef CAS .
-
(a) Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440 CrossRef ;
(b) Z. Yang, G. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038 CrossRef CAS ;
(c) S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070 CrossRef CAS ;
(d) P. K. Vemula, J. Li and G. John, J. Am. Chem. Soc., 2006, 128, 8932 CrossRef ;
(e) R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19 CrossRef CAS ;
(f) Z. Yang, M. Ma and B. Xu, Soft Matter, 2009, 5, 2546 RSC .
-
(a) K. J. C. van Bommel, M. C. Stuart, B. L. Fering and J. van Esch, Org. Biomol. Chem., 2005, 3, 2917 RSC ;
(b) P. K. Vemula, G. A. Cruikshank, J. M. Karp and G. John, Biomaterials, 2009, 30, 383 CrossRef CAS .
- Very recently, a mechanically tough hydrogel formed by supramolecular interactions has been reported: Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339 Search PubMed .
-
(a) M. M. Mohamed and B. F. Sloane, Nat. Rev. Cancer, 2006, 6, 764 CrossRef CAS ;
(b) K. Kose, C. Yazici and O. Ascıoglu, Clin. Biochem., 2001, 34, 125 CrossRef CAS .
-
(a) A. Magklara, A. Scorilas, W. J. Catalona and E. P. Diamandis, Clin. Chem., 1999, 45, 1960 CAS ;
(b) G. Wu, R. H. Datar, K. M. Hansen, T. Thundat, R. J. Cote and A. Majumdar, Nat. Biotechnol., 2001, 19, 856 CrossRef CAS ;
(c) H. Lilja, D. Ulmert and A. J. Vickers, Nat. Rev. Cancer, 2008, 8, 268 CrossRef CAS .
-
(a) M. P. Lutolf, F. E. Weber, H. G. Schmoekel, J. C. Schense, T. Kohler, R. Müller and J. A. Hubbell, Nat. Biotechnol., 2003, 21, 513 CrossRef CAS ;
(b) H. S. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and A. D. Luo, Nat. Mater., 2006, 5, 797 CrossRef CAS ;
(c) A. A. Aimetti, M. W. Tibbitt and K. S. Anseth, Biomacromolecules, 2009, 10, 1484 CrossRef CAS .
- For selected examples, see:
(a) K. L. Niece, J. D. Hartgerink, J. J. J. M. Donners and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 7146 CrossRef CAS ;
(b) L.-s. Li and S. I. Stupp, Angew. Chem., Int. Ed., 2005, 44, 1833 CrossRef CAS ;
(c) M. O. Guler, S. Soukasene, J. F. Hulvat and S. I. Stupp, Nano Lett., 2005, 5, 249 CrossRef CAS ;
(d) Y. Yamamoto, T. Fukushima, A. Saeki, S. Seki, S. Tagawa, N. Ishii and T. Aida, J. Am. Chem. Soc., 2007, 129, 9276 CrossRef CAS ;
(e) Y. Yamamoto, G. Zhang, W. Jin, T. Fukushima, N. Ishii, A. Saeki, S. Seki, S. Tagawa, T. Minari, K. Tsukagoshi and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21051 CrossRef CAS .
- M. Ikeda, Y. Shimizu, S. Matsumoto, H. Komatsu, S.-i. Tamaru, T. Takigawa and I. Hamachi, Macromol. Biosci., 2008, 8, 1019 CrossRef CAS .
-
(a) P. D. Thornton, G. McConnell and R. V. Ulijn, Chem. Commun., 2005, 5913 RSC ;
(b) P. D. Thornton, R. J. Mart and R. V. Ulijn, Adv. Mater., 2007, 19, 1252 CrossRef .
-
(a) S. Kiyonaka, K. Sugiyasu, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 10954 CrossRef CAS ;
(b) S. Kiyonaka, S. Shinkai and I. Hamachi, Chem.–Eur. J., 2003, 9, 976 CrossRef CAS .
-
(a) Y. Zhang, Z. Yang, F. Yuan, H. Gu, P. Gao and B. Xu, J. Am. Chem. Soc., 2004, 126, 15028 CrossRef CAS ;
(b) N. Shi, H. Dong, G. Yin, Z. Xu and S. Li, Adv. Funct. Mater., 2007, 17, 1837 CrossRef CAS .
- Agarose gel (Agarose 900(DOJINDO), 1.0 wt%), which is typically used for DNA gel electrophoresis, shows the yield stress (σc) of 989 Pa on using the same setup for the measurements.
- To prevent evaporation of water and to obtain a spherically-shaped hydrogel capsule easily, sol 1 was incubated in HD.
- The structural stability of the SH-capsule 1 in water depended on the size of the capsule, the concentration of 1, and the presence of buffers and salts; SH-capsules 1 (10 wt%) greater than 2 μL (1680 μm in diameter) were stable in distilled water without significant change of their shapes for 6–24 h, whereas SH-capsules 1 smaller than 1 μL (1090 μm in diameter) shrank significantly within 6 h (ESI†, Fig. S2).
-
(a) R. Matzke, K. Jacobson and M. Radmacher, Nat. Cell Biol., 2001, 3, 607 CrossRef CAS ;
(b) H. Zhang, E. Tumarkin, R. Peerani, Z. Nie, R. M. A. Sullan, G. C. Walker and E. Kumacheva, J. Am. Chem. Soc., 2006, 128, 12205 CrossRef CAS .
-
(a) H. Haga, S. Sasaki, M. Morimoto, K. Kawabata, E. Ito, K. Abe and T. Sambongi, Jpn. J. Appl. Phys., 1998, 37, 3860 CrossRef CAS ;
(b) M. Stolz, R. Raiteri, A. U. Daniels, M. R. VanLandingham, W. Baschong and U. Aebi, Biophys. J., 2004, 86, 3269 CrossRef CAS .
- The Higuchi square root time model27 is the most widely used one to describe drug release from matrices, which is derived from a planar matrix but is applicable for different shaped matrices.
-
(a) T. Higuchi, J. Pharm. Sci., 1963, 52, 1145 CrossRef CAS ;
(b) P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 23 CrossRef CAS ;
(c) C.-C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379 CrossRef CAS .
- Rapid burst release of pyrene butyric acid observed at an early stage could be described as due to a small amount of pyrene butyric acid remaining at the interface between the SH-capsule 1 and HD.
- L. Ruan, H. Zhang, H. Luo, J. Liu, F. Tang, Y.-K. Shi and X. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5105 CrossRef CAS .
- J. M. Fitzpatrick, R. S. Kirby, C. L. Brough and A. L. Saggerson, Prostate Cancer Prostatic Dis., 2009, 12, 347 Search PubMed .
- DUPA shows a high affinity toward purified PSMA (Ki = 8 nM) and could accept a wide range of substituents at the C2 position with minimal loss of affinity32.
- A. A. P. Kozikowski, J. Zhang, F. Nan, P. A. Petukhov, E. Grajkowska, J. T. Wroblewski, T. Yamamoto, T. Bzdega, B. Wroblewska and J. H. Neale, J. Med. Chem., 2004, 47, 1729 CrossRef CAS .
- S. Rehault, M. Brillard-Bourdet, L. Bourgeois, G. Frenette, L. Juliano, F. Gauthier and T. Moreau, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2002, 1596, 55 Search PubMed .
-
(a) E. H. Holmes, T. G. Greene, W. T. Tino, A. L. Boynton, H. C. Aldape, S. L. Misrock and G. P. Murphy, Prostate, 1996, 29, 25 CrossRef ;
(b) M. Kawakami and J. Nakayama, Cancer Res., 1997, 57, 2321 CAS ;
(c) A. Ghosh and W. D. Heston, J. Cell. Biochem., 2004, 91, 528 CrossRef CAS .
- H. Liu, A. K. Rajasekaran and P. Moy, Cancer Res., 1998, 58, 4055 CAS .
- Loading efficiency of 2 into the SH-capsule 1 was evaluated to be 89, 95, 94% for 1, 2, 4 μL of capsules, respectively (ESI†, Fig. S5). A leakage of 2 from the SH-capsule 1/2 was observed at 37 °C (25% at 360 min (loading efficiency = 75%, ESI†, Fig. S6). However, this result suggests that the release function can also be controlled by thermal gel-to-sol transition of the SH-capsule 1.
- A fragment 3 (calcd. for [M]+: m/z = 1458.6; found: 1459.3) was detected by MALDI TOF MS of the supernatant. The release speed was almost independent of the size of the capsules tested in this study; release % at 360 min was evaluated to be 65, 49, 48% for 1, 2, 4 μL of capsules, respectively (ESI†, Fig. S5).
- H. Koistinen, G. Wohlfahrt, J. M. Mattsson, P. Wu, J. Lahdenperä and U.-H. Stenman, Prostate, 2008, 68, 1143 CrossRef CAS .
-
(a) B. Paul, R. Dhir, D. Landsittel, M. R. Hitchens and R. H. Getzenberg, Cancer Res., 2005, 65, 4097 CrossRef CAS ;
(b) B. Acevedo, Y. Perera, M. Ruiz, G. Rojas, J. Benítez, M. Ayala and J. Gavilondo, Clin. Chim. Acta, 2002, 317, 55 CrossRef CAS .
- For instance, it is reported that Hep G2 cells secrete plasma proteins including various enzymes such as proteases and lipases41.
-
(a) B. G. Grimwood, T. H. Plummer, Jr and A. L. Tarentino, J. Biol. Chem., 1989, 264, 15662 CAS ;
(b) C. Ching-Hong, T. H. Forte, B. E. Cahoon, R. N. Thrift and J. J. Albers, Biochim. Biophys. Acta, Lipids Lipid Metab., 1986, 877, 433 Search PubMed .
-
(a) B. S. Slusher, J. J. Vornov, A. G. Thomas, P. D. Hurn, I. Harukuni, A. Bhardwaj, R. J. Traystman, M. B. Robinson, P. Britton, X.-C. May Lu, F. C. Tortella, K. M. Wozniak, M. Yudkoff, B. M. Potter and P. F. Jackson, Nat. Med., 1999, 5, 1396 CrossRef CAS ;
(b) P. F. Jackson and B. S. Slusher, Curr. Med. Chem., 2001, 8, 949 CAS .
-
(a) S. A. Kularatne, K. Wang, H.-K. R. Santhapuram, P. S. Philip and S. Low, Mol. Pharmaceutics, 2009, 6, 780 CrossRef CAS ;
(b) R. P. Murelli, A. X. Zhang, J. Michel, W. L. Jorgensen and D. A. Spiegel, J. Am. Chem. Soc., 2009, 131, 17090 CrossRef CAS .
- An increase in the fluorescence intensity coming from TAMRA of the fragment 3 around LNCAP was also observed in the presence of PMPA (ESI†, Fig. S10).
Footnote |
| † Electronic supplementary information (ESI) available: Molecular structures of BODIPY, PMPA, and PSA inhibitor. Synthetic procedures of 5 and TAMRA-K(DUPA)FSSIYSQTEEQC-CONH2 peptide. Fig. S1–S11. See DOI: 10.1039/c0sc00278j |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.