Catalytic asymmetric hydrophosphonylation of ynones†
Daisuke
Uraguchi
,
Takaki
Ito
,
Shinji
Nakamura
and
Takashi
Ooi
*
Department of Applied Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho B2-3(611), Chikusa, Nagoya 464-8603, Japan. E-mail: tooi@apchem.nagoya-u.ac.jp; Fax: +81-(0)52-789-3338; Tel: +81-(0)52-789-4501
First published on 24th June 2010
Abstract
Catalytic, highly enantioselective hydrophosphonylation of ynones has been achieved by exploiting the characteristic features of in situ generated chiral tetraaminophosphonium phosphite.
Since chiral α-hydroxy phosphonates and phosphonic acids exert intriguing activities as bio-phosphate mimics, antibiotics, antivirals, and antitumor agents, they have stimulated enormous synthetic studies over the past several decades.1 The enantioselective addition of dialkyl phosphonate to a carbonyl compound, i.e., hydrophosphonylation, is one of the most straightforward processes for the stereoselective construction of phosphorus–carbon (P–C) bonds in organic chemistry, providing a practical access to chiral α-hydroxy phosphonates.2 So far, several efficient catalytic methods have been elaborated for this transformation, mainly involving the use of aldehydes as a substrate.3,4 In marked contrast, however, a reliable catalytic system for coupling simple ketones and dialkyl phosphonates to form α-tetrasubstituted α-hydroxy phosphonates, which may potentially exhibit increased resistance to protease enzymes and enhanced bioactivity, has scarcely been developed even with an achiral catalyst.5–8 This methodological deficiency could stem from the following intrinsic problems: (1) the low electrophilicity of ketone carbonyls; (2) the predominant contribution of the less reactive phosphonate form to the equilibrium in phosphonate–phosphite tautomerism;9 and (3) the problematic and unavoidable retro-hydrophosphonylation and phospha-Brook rearrangement in typically employed, base-catalyzed systems (Scheme 1).10,11 For establishing absolute stereochemical control, difficulty in differentiating the two substituents on the prochiral ketone carbon appears to be an additional obstacle. Quite recently, Feng and co-workers reported the effectiveness of Lewis acids such as titanium isopropoxide in facilitating the hydrophosphonylation of ketones under solvent-free conditions. They also succeeded in obtaining moderate enantioselectivity in a preliminary experiment using a chiral tridentate ligand.7 Although these situations with the catalytic asymmetric hydrophosphonylation of simple ketones seem to emphasize the difficulties associated with developing efficient chiral organic base catalysts, we were interested in the possibility of offering our own solution to this formidable synthetic challenge by overcoming the long-standing reactivity and selectivity issues in the context of our research aimed at the design and synthetic applications of chiral tetraaminophosphonium salts.4,12
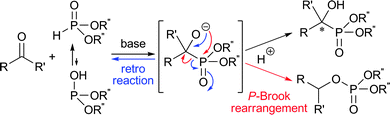 | ||
| Scheme 1 Possible side reactions of hydrophosphonylation of ketones under basic conditions. | ||
We recently developed a highly efficient, catalytic asymmetric hydrophosphonylation of aldehydes by the utilization of catalytically generated tetraaminophosphonium phosphite A (Scheme 2) as a uniquely reactive chiral P-nucleophile.4 The key features to manipulate this unstable organic ion pair are the double hydrogen-bonding capability of the chiral cation of aminophosphonium chloride 1 and the strong basicity of its conjugate base, triaminoiminophosphorane 2. On the basis of these findings, we assumed that the phosphite anion with extremely high nucleophilicity could participate in facile bond formation with less reactive ketones stereoselectively. Furthermore, after the addition to ketones, the rapid proton transfer from the weakly acidic N–H proton of the aminophosphonium cation to the intermediary alkoxide anion would occur in a pseudo-intramolecular manner (B), which could prevent both retro-hydrophosphonylation and phospha-Brook rearrangement, thereby yielding the desired α-tetrasubstituted α-hydroxy phosphonates as the sole products.13 Herein, we describe the realization of this hypothesis in achieving highly enantioselective hydrophosphonylation of ynones mediated by P-spiro chiral tetraaminophosphonium chloride 1d (see Table 2).
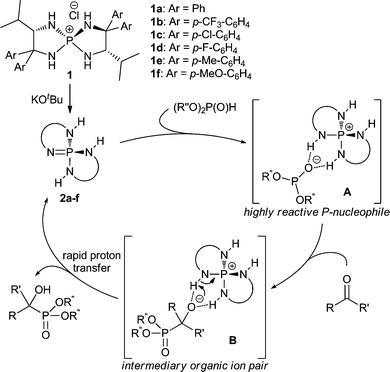 | ||
| Scheme 2 Working hypothesis. | ||
We decided to examine the hydrophosphonylation of ynones with consideration of the vast synthetic utilities of optically active propargylic alcohols based on the rich chemistry of carbon–carbon triple bonds that can be variably converted to other useful functional groups.14–16 Initially, we attempted the reaction of 3-decyn-2-one (3a) with dimethylphosphite under the influence of triaminoiminophosphorane 2a prepared in situ from aminophosphonium salt 1a and KOtBu in THF at −78 °C. As expected, the P–C bond formation took place cleanly and reached completion in 24 h, furnishing the desired α-tetrasubstituted α-hydroxy phosphonate 4a in quantitative isolated yield with 83% ee (entry 1, Table 1). Then, we evaluated the effect of an aromatic substituent on 1 (Ar) on the catalytic and chiral efficiencies, which revealed the importance of a balance between its electronic properties and steric demand (entries 2–6). When the reaction was performed with 1b bearing a strongly electronegative and relatively bulky p-trifluoromethylphenyl moiety, both the chemical yield and stereoselectivity were diminished (entry 2). Interestingly, the introduction of p-mono-halogenated aromatic groups turned out to be beneficial, and the highest enantioselectivity was attained by employing p-fluorophenyl-substituted 1d (entries 3 and 4). It should be noted that the use of electron-rich aminophosphonium cations such as 1e and 1f decreased the enantiomeric excess (entries 5 and 6).
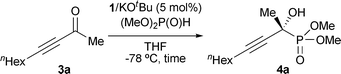
The optimal catalyst precursor, chiral aminophosphonium chloride 1d, was used for further investigation to probe the scope of the ynone component with respect to the substituent at the alkyne terminus. The representative results summarized in Table 2 indicate the applicability of the present system. With 5 mol% of iminophosphorane 2d generated in situ from 1d/KOtBu, the hydrophosphonylation is generally completed within 22 h to afford the corresponding α-hydroxy phosphonate 4 in excellent chemical yield with a high level of enantioselectivity, except for the case of 3-hexyn-2-one (3b) (entry 1). Ynones possessing a long linear chain or an α-branched alkyl group served as a good substrate (entries 2 and 3), while the incorporation of a β-branched alkyl substituent led to lower enantioselectivity (entry 4). Alkoxy and silyloxy functionalities were tolerated, where the steric effect of the appendages on the reactivity and selectivity appeared to be marginal (entries 5–7). In addition, the reaction with trimethylsilyl-substituted ynone 3i proceeded similarly to give 4i in 96% yield with 91% ee (entry 8).‡
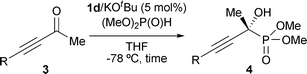
| Entry | R | 3 | Time/h | Yield (%)b | ee (%)c | 4 |
|---|---|---|---|---|---|---|
| a See ESI for details. b Isolated yield. c Enantiomeric excess was analyzed by chiral HPLC. Absolute configurations of 4b–h were assigned by analogy to 4i. d The product 4h was isolated after removal of Me3Si group. e The absolute configuration of 4i was determined by X-ray analysis. | ||||||
| 1 | Et | 3b | 76 | 73 | 90 | 4b |
| 2 | Me(CH2)8 | 3c | 22 | 98 | 88 | 4c |
| 3 | Cyclohexyl | 3d | 17 | 96 | 90 | 4d |
| 4 | Me2CHCH2 | 3e | 16 | 97 | 79 | 4e |
| 5 | BnOCH2 | 3f | 17 | 97 | 81 | 4f |
| 6 | t BuMe2SiO(CH2)2 | 3g | 19 | 96 | 87 | 4g |
| 7 | Me3SiO(Me)2C | 3h | 19 | 96 | 88 | 4h |
| 8 | Me3Si | 3i | 17 | 96 | 91 | 4i |
In conclusion, we have achieved the first highly enantioselective hydrophosphonylation of ynones by exploiting the eminent nucleophilicity of tetraaminophosphonium phosphite as well as the proton-donating and stereocontrolling abilities of the chiral aminophosphonium cation. This study not only provides a new synthetic route to optically active α-tetrasubstituted α-hydroxy phosphonates but also highlights the potential of chiral triaminoiminophosphorane as a strong organic base catalyst with hydrogen-bonding donor capability.
Acknowledgements
This work was supported by Grants of JSPS for Scientific Research and the Global COE program in Chemistry of Nagoya University.Notes and references
- For reviews, see: R. Engel, Org. React., 1988, 36, 175 Search PubMed; A Guide to Organophosphorus Chemistry, ed. L. D. Quin, John Wiley & Sons, New York, 2000 CAS; O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2005, 16, 3295 CAS.
- For reviews on hydrophosphonylation, see: A. N. Pudovik and I. V. Konovalova, Synthesis, 1979, 81 Search PubMed; D. Enders, A. Saint-Dizier, M.-I. Lannou and A. Lenzen, Eur. J. Org. Chem., 2006, 29 CrossRef CAS; P. Merino, E. Marqués-López and R. P. Herrera, Adv. Synth. Catal., 2008, 350, 1195 CrossRef CAS; Ł. Albrecht, A. Albrecht, H. Krawczyk and K. A. Jørgensen, Chem.–Eur. J., 2010, 16, 28 CrossRef CAS; S. Sobhani and Z. Tashrifi, Tetrahedron, 2010, 66, 1429 CrossRef CAS.
- H. Wynberg and A. A. Smaardijk, Tetrahedron Lett., 1983, 24, 5899 CrossRef CAS; A. A. Smaardijk, S. Noorda, F. van Bolhuis and H. Wynberg, Tetrahedron Lett., 1985, 26, 493 CrossRef CAS; T. Yokomatsu, T. Yamagishi and S. Shibuya, Tetrahedron: Asymmetry, 1993, 4, 1779 CrossRef CAS; T. Arai, M. Bougauchi, H. Sasai and M. Shibasaki, J. Org. Chem., 1996, 61, 2926 CrossRef CAS; M. D. Groaning, B. J. Rowe and C. D. Spilling, Tetrahedron Lett., 1998, 39, 5485 CrossRef CAS; J. P. Duxbury, A. Cawley, M. Thornton-Pett, L. Wantz, J. N. D. Warne, R. Greatrex, D. Brown and T. P. Kee, Tetrahedron Lett., 1999, 40, 4403 CrossRef CAS; T. D. Nixon, S. Dalgarno, C. V. Ward, M. Jiang, M. A. Halcrow, C. Kilner, M. Thornton-Pett and T. P. Kee, C. R. Chim., 2004, 7, 809 CrossRef CAS; B. Saito, H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 1978 CrossRef CAS; P. Merino, E. Marqués-López and R. P. Herrera, Adv. Synth. Catal., 2008, 350, 1195 CrossRef CAS; X. Zhou, X. Liu, X. Yang, D. Shang, J. Xin and X. Feng, Angew. Chem., Int. Ed., 2008, 47, 392 CrossRef CAS; S. Gou, X. Zhou, J. Wang, X. Liu and X. Feng, Tetrahedron, 2008, 64, 2864 CrossRef CAS; J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 10521 CrossRef CAS; F. Yang, D. Zhao, J. Lan, P. Xi, L. Yang, S. Xiang and J. You, Angew. Chem., Int. Ed., 2008, 47, 5646 CrossRef CAS; K. Suyama, Y. Sakai, K. Matsumoto, B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2010, 49, 797 CAS.
- D. Uraguchi, T. Ito and T. Ooi, J. Am. Chem. Soc., 2009, 131, 3836 CrossRef CAS.
- For catalytic asymmetric approaches to α-tetrasubstituted α-hydroxy phosphonates via C–C bond formation, see: S. Samanta and C.-G. Zhao, J. Am. Chem. Soc., 2006, 128, 7442 Search PubMed; T. Mandal, S. Samanta and C.-G. Zhao, Org. Lett., 2007, 9, 943 CrossRef CAS; J. Liu, Z. Yang, Z. Wang, F. Wang, X. Chen, X. Liu, X. Feng, Z. Su and C. Hu, J. Am. Chem. Soc., 2008, 130, 5654 CrossRef CAS; X. Chen, J. Wang, Y. Zhu, D. Shang, B. Gao, X. Liu, X. Feng, Z. Su and C. Hu, Chem.–Eur. J., 2008, 14, 10896 CrossRef CAS.
- For hydrophosphonylation to highly activated ketones, see: F. Wang, X. Liu, X. Cui, Y. Xiong, X. Zhou and X. Feng, Chem.–Eur. J., 2009, 15, 589 Search PubMed.
- X. Zhou, Y. Liu, L. Chang, J. Zhao, D. Shang, X. Liu, L. Lin and X. Feng, Adv. Synth. Catal., 2009, 351, 2567 CrossRef CAS.
- A. N. Pudovik and O. S. Durova, Zh. Obshch. Khim., 1966, 36, 1465; A. N. Pudovik and O. S. Shulyndi, Zh. Obshch. Khim., 1968, 38, 2009; M. Y. Dvorko, T. E. Glotova, I. A. Ushakov, N. K. Gusarova and B. A. Trofimov, Russ. J. Org. Chem., 2008, 44, 1245 CrossRef CAS.
- G. O. Doak and L. D. Freedman, Chem. Rev., 1961, 61, 31 CrossRef CAS; W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 3594 CrossRef CAS.
- M. S. Kharasch, R. A. Mosher and I. S. Bengelsdorf, J. Org. Chem., 1960, 25, 1000 CrossRef CAS; M. Sekine, M. Nakajima, A. Kume, A. Hashizume and T. Hata, Bull. Chem. Soc. Jpn., 1982, 55, 224 CAS; E. Öhler, M. El-Badawi and E. Zbiral, Chem. Ber., 1985, 118, 4099 CrossRef; R. Gancarz, I. Gancarz and U. Walkowiak, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 104, 45 Search PubMed; H. Maeda, K. Takahashi and H. Ohmori, Tetrahedron, 1998, 54, 12233 CrossRef CAS.
- H. Timmler and J. Kurz, Chem. Ber., 1971, 104, 3740 CrossRef CAS; F. Hammerschmidt, E. Schneyder and E. Zbiral, Chem. Ber., 1980, 113, 3891; B. A. Arbuzov, A. V. Fuzhenkova and N. A. Banderova, Zh. Obshch. Khim., 1986, 56, 1691 CAS; F. Hammerschmidt, Monatsh. Chem., 1993, 124, 1063 CrossRef CAS; L. El Kaïm, L. Gaultier, L. Grimaud and A. Dos Santos, Synlett, 2005, 2335 CrossRef CAS.
- D. Uraguchi, S. Sakaki and T. Ooi, J. Am. Chem. Soc., 2007, 129, 12392 CrossRef CAS; D. Uraguchi, T. Ito, S. Nakamura, S. Sakaki and T. Ooi, Chem. Lett., 2009, 38, 1052 CrossRef CAS.
- Although intermediary ion-pair B could not be identified, the regeneration of iminophosphorane 2a through the formation of product 4a was confirmed by 31P NMR analysis at −98 °C in THF.
- Modern Acetylene Chemistry, ed. P. J. Stang and F. Diederich, VCH, Weinheim, 1995 Search PubMed.
- For synthetic utilities of alkynyl hydroxy phosphonate, see: F. Benayoud, D. J. deMendonca, C. A. Digits, G. A. Moniz, T. C. Sanders and G. B. Hammond, J. Org. Chem., 1996, 61, 5159 Search PubMed.
- Hydrophosphonylation of simple aromatic ketones such as acetophenone with 1a/KOtBu cleanly gave the corresponding product, albeit with low to moderate enantioselectivity.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of 1, 3, and 4. CCDC reference number 772834. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00268b |
| ‡ Crystal data for 4i (CCDC 772834): C9H19O4PSi (Mw 250.30), monoclinic, P21, a = 9.5228(13), b = 7.4316(11), c = 9.7945(14) Å, β = 103.092(3)°, V = 675.14(17) Å3, Z = 2, T = 153(2) K, independent reflections 3033 [Rint = 0.0248], Flack parameter 0.17(10), R1(Rw) = 0.0388 (0.0968) (I > 2σ(I)), GOF = 1.071. |
| This journal is © The Royal Society of Chemistry 2010 |