Guest-responsive porous magnetic frameworks using polycyanometallates†
Masaaki
Ohba
*ab,
Ko
Yoneda
a and
Susumu
Kitagawa
*abc
aDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: ohba@sbchem.kyoto-u.ac.jp; Fax: +81-75-383-2732; Tel: +81-75-383-2807
bRIKEN Spring-8 Center, Kouto, Sayo-cho, Sayo-gun, Hyogo, 679-5198, Japan
cInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto, 606-8501, Japan. E-mail: kitagawa@sbchem.kyoto-u.ac.jp
First published on 7th October 2009
Abstract
Three types of cyanide-bridged porous magnetic frameworks based on polycyanometallates exhibited reversible magnetic conversions associated with different guest-responsive structural transformation, which highlights the available strategy using a polycyanometallate for constructing the guest-responsive magnetic frameworks.
Introduction
Molecular-based magnets (MMs) have been studied extensively since the end of the 1980s.1,2 These studies started with the challenge to achieve long-range magnetic ordering with controlling magnetic interactions and arrangement of magnetic centres. Several functionalized MMs, e.g., magnetic switches in response to light,3 pressure,4 solvent5etc., have been developed through various trials. These efforts paved the way towards new magnetic materials, and developed into a new active research area.7 The basic molecular design for MMs with infinite structures has aspects in common with that of coordination polymers (CPs) or metal–organic frameworks (MOFs) that have frameworks extended by coordination bonds.8 Thus, MMs and CPs providing magnetic properties can be identified as coordination polymer magnets (CPMs). As is obvious, CPMs inherit the characteristics of CPs, such as high regularity, diversity, and flexibility in combining a wide range of components, e.g., bridging ligands, metal ions, co-ligands, etc., with elaborately designed frameworks. In particular, the flexible frameworks have the potential to respond to external physical and chemical stimuli. Herein, we focus on the design of CPMs to implement guest-responsive magnetic switching.The porous framework is one noteworthy functionalized architecture derived from CPs. In the past decade, porous coordination polymers (PCPs) have emerged as a new class of porous materials, representing an evolved form of CPs.8 Flexible PCPs' pores allow expansion or shrinkage of their framework in association with guest adsorption or desorption.9 Therefore, PCPs are expected to be multifunctional platforms exhibiting porous properties with specific chemical and physical properties. From this perspective, porous coordination polymer magnets (PCPMs) incorporating magnetic properties into the framework would be ideal multifunctional materials, particularly as chemoresponsive materials.6a,b Kahn et al. first introduced the concept of a “magnetic sponge” in molecular magnets that can reversibly release and reabsorb both non-coordinated and coordinated solvent molecules, accompanied by a drastic change of magnetic properties.5a,b Although this concept restricts the intended guest molecules to solvents in non-porous systems, it expresses the essence of guest-responsivity in PCPMs. Here we use “guest-responsive magnets” as an umbrella term instead of magnetic sponges.
To couple the magnetic and porous properties, we need a more sophisticated design strategy. Magnetic ordering requires short bridging structures to mediate strong magnetic interactions between magnetic centres, while porous frameworks generally consist of relatively long bridging ligands forming a large inner space. One rational design for PCPMs is based on using large paramagnetic compounds (linkers) to combine magnetic centres. From this viewpoint, a polycyanometallate anion, [MA(CN)n]m− (n = 2–8), is a suitable linker because it gives a relatively long MB–NC–MA–CN–MB linkage (ca. 1 nm) for the inner pore and mediates effective magnetic interaction between MA and MB (Scheme 1). Prussian Blue, FeIII4[FeII(CN)6]3·nH2O, is representative of magnetic compounds using a polycyanometallate linker. A number of Prussian Blue analogues (PBAs) have been reported so far.10 Several PBAs exhibit magnetic and porous properties, but the cyanide bridges do not perform structural flexibility except for structural defects because of their close-packed structures.6a,11 A convertible structure responsive to chemical stimuli is a significant mechanism to develop guest-responsive magnets. The key to creating such a flexible but durable framework is to utilize weak molecular interactions in addition to strong coordination bonds. From this perspective, we have extensively studied cyanide-bridged CPMs by the use of [MA(CN)n]m− and secondary building units (SBUs) consisting of metal ions MBn+ and auxiliary ligands (co-ligands).4e,f, 6h,i, 12,13 The co-ligands play a significant role in avoiding the formation of a PB-type structure and deriving the desired frameworks based on rational molecular designs by limiting the available coordination sites on the SBU. In addition, the incorporation of co-ligands prevents formation of close-packed structures and results in non-bridging cyano groups in the framework. The non-bridging cyano groups act as guest-interactive sites by forming hydrogen bonds, which is of considerable importance for reversible structural conversion.
 | ||
| Scheme 1 Major characteristics of the polycyanometallate anion | ||
In this paper, we highlight our strategy for constructing guest-responsive magnets based on the utility component, polycyanometallate, with three types of guest-responsive cyanide-bridged PCPMs, i.e.: (1) reversible magnetic and structural conversion of [Ni(dipn)]2[Ni(dipn)(H2O)][Fe(CN)6]2·11H2O (1: dipn = N,N-di(3-aminopropyl)amine)6h triggered by water desorption/adsorption; (2) reversible topochemical transformation of a ferrimagnet, [Mn(HL)(H2O)][Cr(CN)6]·H2O (2: L = N,N-dimethylethylenediamine);6i and (3) bidirectional magnetic chemoswitching of {Fe(pz)[Pt(CN)4]} (3: pz = pyrazine).13
Experimental
Physical measurements
Variable-temperature X-ray powder diffraction was carried out on a Rigaku RINT-2000 Ultima diffractometer with Cu Kα radiation. The magnetic susceptibilities of all samples were measured on a Quantum Design MPMS – XL5R SQUID susceptometer in the temperature range 2–300 K in an applied dc field of 500 Oe. The samples were placed in a glass tube and fixed to the end of the sample transport rod. Dehydrated samples were prepared by vacuuming for 2 h in the SQUID sample chamber at 400 K. The guest molecules were injected into the glass tube through the sample transport rod with controlling its vapor pressure at 293 K. The molar magnetic susceptibility, χM, was corrected for the diamagnetism of the constituent atoms.X-Ray diffraction data of 1, 2 and 2a were collected on a Rigaku Mercury CCD system while 3(HS) and 3(LS) were collected on a Rigaku Varimax CCD system. In all cases, graphite-monochromated Mo Kα radiation (λ = 0.71070 Å) was used. A single crystal was mounted on a fiber loop with liquid paraffin and the temperature kept constant under flowing N2. The crystal parameters are listed in Table 1. All structures were solved by a standard direct method and expanded using Fourier techniques. Full-matrix least-squares refinements were carried out using Crystal Clear 1.4. (crystallographic software package of the Molecular Structure Corp. and Rigaku) for 1, 3(HS) and 3(LS) and teXsan (crystal structure analysis package of the Molecular Structure Corp.) for 2 and 2a, with anisotropic thermal parameters for all non-hydrogen atoms. All of the hydrogen atoms were placed in the calculated positions and refined using a riding model.
| Compound | 1 | 2 | 2a | 3(HS) | 3(LS) |
|---|---|---|---|---|---|
| a The structures of 2 and 2a were re-examined using newly-processed diffraction data. | |||||
| Formula | C30H63N31Fe2Ni3O11 | C10H17CrMnN8O2 | C10H13CrMnN8 | C8H8N6FeO2Pt | C8H8N6FeO2Pt |
| Formula weight | 1209.86 | 388.23 | 352.20 | 471.11 | 471.11 |
| No. of water | 11 | 2 | 0 | 2 | 2 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Tetragonal | Tetragonal |
| Space group | C2/c | P21/n | P21/c | P4/mmm | P4/mmm |
| T/K | 243 | 243 | 323 | 293 | 293 |
| a/Å | 24.044(8) | 7.680(3) | 7.84(2) | 7.457(4) | 7.184(6) |
| b/Å | 14.343(4) | 14.499(5) | 14.31(3) | 7.457(4) | 7.184(6) |
| c/Å | 16.688(5) | 16.596(5) | 12.96(2) | 7.259(4) | 6.783(5) |
| β/° | 100.552(4) | 97.323(5) | 90.07(3) | 90 | 90 |
| V/Å3 | 5658(3) | 1833.0(10) | 1455.6(4) | 403.6(4) | 350.1(5) |
| Z | 4 | 4 | 4 | 1 | 1 |
| µ/mm−1 | 1.547 | 1.298 | 1.616 | 9.503 | 10.957 |
| No. of total data | 21405 | 4294 | 3173 | 3697 | 2796 |
| No. of unique data | 6442 | 4134 | 3046 | 412 | 278 |
| No. of data (I > 2σ(I)) | 4745 | 2333 | 1020 | 344 | 266 |
| R int | 0.0628 | 0.0647 | 0.0984 | 0.0472 | 0.0494 |
| R (all data) | 0.0833 | 0.0947 | 0.1567 | 0.0597 | 0.0512 |
| Rw (all data) | 0.2049 | 0.0955 | 0.1368 | 0.0844 | 0.0737 |
| D c/g cm−3 | 1.420 | 1.407 | 1.607 | 1.938 | 2.234 |
| D c /g cm−3 (without lattice water) | 1.188 | 1.341 | 1.607 | 1.790 | 2.064 |
| Solvent-accessible void/% | 28.2 | 5.2 | 0.0 | 22.4 | 18.1 |
| Ref. | 6h | 6i | 6i | 13 | 13 |
[Ni(dipn)]2[Ni(dipn)(H2O)][Fe(CN)6]2·11H2O (1)
Compound 1 was obtained as black crystals by the reaction of NiCl2·6H2O, dipn and K3[Fe(CN)6] in a 3:3:2 mole ratio in DMF–water solution.6h The asymmetric unit of 1 consists of one [Fe(CN)6]3− anion, one [Ni1(dipn)]2+ and one half [Ni2(dipn)(H2O)]2+ cations and 5.5 water molecules. In [Ni2(dipn)(H2O)]2+ unit, Ni2 and central N atom the dipn lies on a twofold axis. In the lattice, this compound forms a 3-D porous framework consisting of 2-D layers extended by an alternating array of [Ni1(dipn)]2+ and [Fe(CN)6]3−, and [Ni2(dipn)(H2O)]2+ units linking the 2-D layers (Fig. 1). The porous framework has a honeycomb-type channel structure with a solvent-accessible void of 29.0%. The tridentate co-ligand, dipn, coordinates to Ni2+ in meridional positions and gives three coordinable sites on the SBU, [Ni(dipn)]2+. Four cyano groups of [Fe(CN)6]3− coordinate to the adjacent Ni2+ ions, and the other two in cis positions are non-bridging and face the pore. Most of the lattice water molecules are disordered and reside in the pore. In the asymmetric unit, only one lattice water molecule is immobilized by forming bidirectional hydrogen bonds with two non-bridging cyano groups (Fig. 2). | ||
| Fig. 1 Projection of 1 onto the ab plane. Disordered lattice water molecules are omitted. Atoms: Fe (orange), Ni (green), O (red), N (light blue), C (gray). | ||
 | ||
| Fig. 2 Bidirectional hydrogen bonds (pink dotted lines) between lattice water molecules and non-bridging cyano groups. | ||
The as-synthesized compound 1 exhibited ferromagnetic ordering at 8.5 K based on the strict orthogonality of magnetic orbitals of Ni2+ and low-spin Fe3+. After vacuum treatment at room temperature, a dehydrated form, [Ni(dipn)]2[Ni(dipn)(H2O)][Fe(CN)6]2·H2O (1a), was obtained and this form showed amorphous-like XRPD patterns and paramagnetic behaviour (Fig. 3). The dehydrated form 1a adsorbed water at room temperature with a maximum uptake of 9.93 mol/mol. The rehydrated form (1b) showed the same XRPD pattern and magnetic behaviour of the initial form 1 (Fig. 3). The reversible crystal-to-amorphous-like phase transformation could be performed repeatedly by water desorption/adsorption processes. In contrast, the anhydrous form, [Ni(dipn)]3[Fe(CN)6]2 (1c), was an amorphous paramagnet and did not recover the initial crystallinity and ferromagnetic phase upon exposure to gaseous water or immersion in water.
 | ||
Fig. 3 Temperature dependences of χM for the initial form 1 (●), the dehydrated form 1a ( ), the rehydrated form 1b ( ), the rehydrated form 1b ( ) and the anhydrous form 1c (□) in an applied dc field of 500 Oe. The insert shows the corresponding XRPD patterns. ) and the anhydrous form 1c (□) in an applied dc field of 500 Oe. The insert shows the corresponding XRPD patterns. | ||
The flexible Fe–CN–Ni linkage, bidirectional hydrogen bonds between lattice water molecules and non-bridging cyano nitrogen atoms play crucial roles in the reversible structural and magnetic conversion. Other porous cyanide-bridged magnetic frameworks, e.g., Cu3[W(CN)8](pym)2·8H2O (pym = pyrimidine),6j {[Mn(HL)(H2O)]2Mn[Mo(CN)7]2}·2H2O (L = N,N-dimethylalaninol)6l and {[Ni(cyclam)]3[W(CN)8]2}·16H2O (cyclam = 1,4,8,11-tetraazacyclotetradecane)6m have been reported, and showed both porous and magnetic properties. These results demonstrate the viability of our strategy of using a polycyanometallate and co-ligands for the construction of porous magnetic frameworks.
[Mn(HL)(H2O)][Cr(CN)6]·H2O (2)
Compound 2 was prepared as pale-green crystals by reaction of K3[Cr(CN)6], MnCl2·4H2O and co-ligand L (NNdmen) in a deoxygenated aqueous solution at room temperature.6i The asymmetric unit of 2 consists of [Mn(HL)(H2O)]3+ cation, [Cr(CN)6]3− anion and a lattice water molecule. In the lattice the compound forms a 2-D layer structure extended by Cr–CN–Mn linkages (Fig. 4a). Four cyano nitrogen atoms in the equatorial positions of [Cr(CN)6]3− coordinate to adjacent Mn2+ ions, and the other two in axial positions, are non-bridging. One protonated co-ligand and one water molecule are located at the axial positions of the Mn2+ ion as monodentate ligands. The coordinated water molecules form Mn–OH2⋯NC–Cr hydrogen bonds with non-bridging cyano groups in the next layer (Fig. 5a). The hydrogen bonds adjust the topology between layers. A dehydrated form, [Mn(HL)][Cr(CN)6] (2a), was obtained as a single crystal by careful heating of 2 at 343 K. After dehydration, the 2-D layers were changed to a 3-D pillared-layer framework, in which the coordinated water molecules were eliminated and extra Cr–CN–Mn bridges were generated between neighbouring sheets by a topochemical reaction (Figs. 4b and 5b). The dehydrated form has a very narrow 1-D channel based on a highly distorted Mn2Cr2 quadrangular gate (0.8 × 1.6 Å2 based on the van der Waals radii).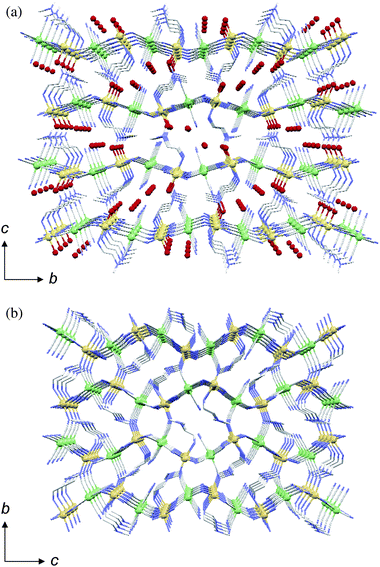 | ||
| Fig. 4 Projections of the initial form 2 onto the bc plane (a) and the dehydrated form 2a onto the ab plane (b). Atoms: Cr (green), Mn (yellow), O (red), N (light blue), C (gray). | ||
 | ||
| Fig. 5 Reversible topotactic structural transformation between 2 (a) and 2a (b). Hydrogen bonds (pink dotted lines). | ||
The initial form exhibited ferrimagnetic ordering with a Tc of 35.2 K, and the Tc was shifted to 60.2 K by dehydration due to increase of the magnetic interaction pathways (Fig. 6). Upon exposure to air or water vapour at 303 K, 2a immediately reproduced the same XRPD pattern and magnetic behavior as 2. The dehydrated form 2a smoothly adsorbed water (2.3 mol/mol) with a large hysteresis because of the restoration of the initial 2-D layer structure, including replacement of the extra cyanide bridges to water molecules. In addition, 2a showed size-selective solvent adsorption, linking chemi- and physisorption processes. The rehydrated form 2b recovered the initial structure and magnetic behaviour of 2. Compound 2 demonstrated a reversible single-crystal-to-single-crystal (SCSC) transformation with shift of Tc through the dehydration/rehydration processes and topochemical reaction.
 | ||
Fig. 6 Temperature dependences of χM for the initial form 2 ( ), the dehydrated form 2a ( ), the dehydrated form 2a ( ) and the rehydrated form 2b ( ) and the rehydrated form 2b ( ) in an applied dc field of 500 Oe. The insert shows the reversible TC switching by dehydration/hydration treatments (red/blue arrows, respectively). ) in an applied dc field of 500 Oe. The insert shows the reversible TC switching by dehydration/hydration treatments (red/blue arrows, respectively). | ||
Analogues using other co-ligands, 1,2-propanediamine (pn)12g,14a and ethylenediamine (en),14b,c also show similar 2-D/3-D conversion with shifting Tc. In addition, conversions between the 0-D paramagnet and 2-D ferromagnet of [K(18-crown-6)(MeOH)2][Mn(L)(H2O)(MeOH)]2[Fe(CN)6]·MeOH (L = N,N′-ethylene-di(5-chlorosalicylideneaminate))5d and between the 1-D metamagnet and 2-D ferromagnet of [Ni(dmen)2]-[Ni(dmen)2(H2O)][Fe(CN)6](bpds)0.5·3H2O (dmen = 1,1-dimethylethylenediamine, bpds2− = 4,4′-biphenyldisulfonate)5e have been reported, based on the same strategy using hexacyanometallates. These compounds provide a flexible magnetic framework incorporating removable solvent co-ligands, and deliver structural and magnetic transformations in response to solvation/desolvation processes. The initial structures are stabilized thermodynamically using solvent co-ligands to avoid forming closed-packed structures. Each independent framework is interlinked to adjacent framework(s) through hydrogen bonds formed between solvent co-ligands and non-bridging cyano groups. The metastable desolvated form is obtained by a topochemical solid-state reaction directed by the hydrogen bonds, and cannot retain its structure under the solvent vapour. Steric repulsion in the desolvated form would be a key driving force to recover the initial structure. From this perspective, the primary building units, [MA(CN)n]m−, are advantageous for constructing guest-responsive (solvato-responsive) magnetic systems, because they: (1) contain extendable and flexible cyanide bridges; (2) are good magnetic mediators; (3) are able to hydrogen bond with protonic solvent molecules; and (4) are moderately bulky generating steric repulsion with co-ligands in adjacent SBUs.
{Fe(pz)[Pt(CN)4]} (3)
Spin crossover (SCO) is a well-known phenomenon in which electron configurations can be switched between high-spin (HS) and low-spin (LS) states in response to external stimuli (temperature, pressure and light irradiation), producing changes in magnetism, colour, dielectric property and structure.3f,13,15 A number of SCO compounds have been elaborated, with control of their spin states in molecules and molecular assemblies, and some have successfully exhibited bistable states around room temperature.16As mentioned above, guest-responsive magnetic conversion in PCPMs and CPMs has been reported but there is a large gap in working temperature between guest adsorption/desorption and the magnetic ordering temperature Tc. The SCO phenomenon would be one solution to overcome this temperature problem instead of ordered magnetism. PCPs incorporating SCO centres (SCO–PCP) may propagate the SCO cooperatively to the whole framework. Such SCO–PCPs may undergo first-order hysteretic spin transition (ST) producing a bistable state at high temperature, in which guest-responsive ST is expected.
Two representative SCO–PCPs, [FeIIL2(NCS)2]·nS (L = 1,2-bis(4-pyridyl)ethylene,17a,btrans-4,4′-azopyridine,17c DL-1,2-bis(4-pyridyl)-1,2-ethanediol;17d S = alcohol molecule), and {Fe(pym)(H2O)[MI(CN)2]2}·H2O,17e have been reported by Real et al. and Kepert et al. These examples clearly display guest-responsive SCO; however, the SCO and guest adsorption–desorption are still not simultaneous. One of the ideal SCO–PCP, {Fe(pz)[Pt(CN)4]} (3), using [Pt(CN)4]2− and pyrazine (pz) co-ligand has been reported by Real et al.13,16b,18 Compound 3 forms a 3-D Hofmann-type framework, in which pz forms rigid bridges between Fe ions as pillars linking the 2-D layers extended by Pt–CN–Fe linkages and forms channels with square Fe2Pt2 windows (Fig. 7). Note that this framework provides two guest-interactive sites: one between the pz bridges (site A) and another between the Pt centres (site B) (Fig. 8). These features should be important a priori for the chemical response of the framework.
 | ||
| Fig. 7 Projection of 3 onto the ac plane. Lattice water molecules are omitted. Atoms: Fe (orange), Pt (pink), N (light blue), C (gray). | ||
 | ||
| Fig. 8 The functional cavity structure of 3. | ||
This compound displays a first-order ST with ca. 25 K-wide hysteresis, with Tc↓ of 285 K and Tc↑ of 309 K. The solvent-accessible voids of the LS and HS states were estimated to be 18.1 and 22.4%, respectively. The guest-free form 3a adsorbs various guest molecules in the gas phase or solution, and forms their clathrates. The spin state changes, depending on the guest molecules. In situ magnetic measurements following guest vapour injection elucidates the simultaneous guest adsorption and bidirectional spin transition (Fig. 9). Most guest molecules transform 3a from the low-spin (LS) state to the high-spin (HS) state, whereas CS2 stabilizes the LS state exceptionally. The stabilized spin states are retained after removing guest molecules, and both spin states are interchangeable by exchanging the guest molecules with controlling vapour pressure.
 | ||
| Fig. 9 Time dependence of the fraction of the HS state under a benzene (yellow) and CS2 (purple) atmosphere at 293 K. Red arrows indicate the starting points of guest injection (valve opening). The color circles are photos of samples. | ||
The guest molecules studied can be grouped into three major classes (Table 2), where class I shows no effect on the spin state; class II stabilizes the HS state; and class III stabilizes the LS state. The structures of pz and CS2 clathrates reveal that: (i) the pz guest molecules are located exclusively at site A; and (ii) the CS2 molecules are located at sites A and B. These systematic structural and magnetic results point to three key factors for stabilizing the spin states: (1) the size and shape of the guest (G); (2) the G⋯pz interaction at site A; and (3) the G⋯Pt interaction at site B. Theoretical calculation of the binding energies between CS2 and each site suggests that the CS2 molecule interacts suitably with both sites, like a key molecule for the framework.13 Compound 3 successfully delivers a desired magnetic switch in response to guest molecules at room temperature. The porous SCO framework is constructed by three functional components; (a) SCO Fe(II) ions as framework joints; (b) flat tetracyanometallate, [M(CN)4]2− (MII = Pt, Pd, Ni), providing guest-interactive sites (site B) and forming flexible M–CN–Fe linkages; and (c) pyrazine co-ligands as rigid pillars and guest-interactive sites (site A) (Fig. 8). The well-designed simple framework provides appropriate interactive pores and allows the spin transition coupled to guest adsorption/desorption processes while retaining its cooperativity.
Conclusions
As shown above, the triad of polycyanometallate, SBU and co-ligand demonstrates the availability for designing and creating guest-responsive magnetic frameworks. In the case of PCPMs 1 and 2, hexacyanometallates work as good linkers and magnetic mediators, but Tc remains in the low temperature region. Guest adsorption and magnetic ordering are not simultaneous events in these compounds. It is necessary to shift the coexistence phase of magnetic and porous properties to a cooperative phase. A more elaborate porous magnetic framework that transcribes a backbone of PBAs with high Tc, e.g., MII3[Cr(CN)6]·nH2O (Tc = 240 K for M = Cr, 315 K for M = V),10 would overcome the temperature problem. Recently, Sutter et al. reported a PCPM, {[Mn(HL)(H2O)]2Mn[Mo(CN)7]2}·2H2O (L = N,N-dimethylalaninol)6l, which provides ring-shaped channels with potential void of 9.2% and exhibits ferrimagnetic ordering at 106 K in a dehydrated form. Such PCPMs with high Tc would exhibit a unique synergy between the magnetic properties and guest molecules, e.g., paramagnetic, chiral or polar guests. On the other hand, the SCO–PCP {Fe(pz)[Pt(CN)4]} (3) notably delivered magnetic chemo-switching at room temperature that will open a route to evolving environmentally responsive materials. Controlling the interactions between the SCO framework and guest molecules will allow precise and universal magnetic switching. As the next step, more advanced functions, e.g., external control of selective guest adsorption, separation, or uptake and release of guest molecules, may be delivered using SCO–PCPs with shape-, volume- and chirality-changeable pores depending on the spin state.Acknowledgements
This work was supported by a ERATO JST Project “Kitagawa Integrated Pore Project”, CREST JST program, Grant-In-Aid for Science Research (B) (No. 20350026) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the Mitsubishi fund.Notes and references
- (a) O. Kahn, Molecular Magnetism, VCH Publishers, 1993 Search PubMed; (b) J. S. Miller and M. Drillon, Magnetism: Molecules to Materials, Wiley-VCH, vol. 2, 2001 Search PubMed; (c) D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS.
- (a) Y. Pei, M. Verdaguer, O. Kahn, J. Sletten and J. P. Renard, J. Am. Chem. Soc., 1986, 108, 7428–7430 CrossRef CAS; (b) J. S. Miller, J. C. Calabrese, H. Rommelmann, S. R. Chittipeddi, J. H. Zhang, W. M. Reiff and A. J. Epstein, J. Am. Chem. Soc., 1987, 109, 769–781 CrossRef CAS; (c) A. Caneschi, D. Gatteschi, R. Sessoli and P. Rey, Acc. Chem. Res., 1989, 22, 392 CrossRef CAS.
- (a) O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 CrossRef CAS; (b) M. Verdaguer, Science, 1996, 272, 698–699 CrossRef CAS; (c) S. I. Ohkoshi and K. Hashimoto, J. Am. Chem. Soc., 1999, 121, 10591–10597 CrossRef CAS; (d) Y. Arimoto, S. I. Ohkoshi, Z. J. Zhong, H. Seino, Y. Mizobe and K. Hashimoto, J. Am. Chem. Soc., 2003, 125, 9240–9241 CrossRef CAS; (e) S. I. Ohkoshi, H. Tokoro and K. Hashimoto, Coord. Chem. Rev., 2005, 249, 1830–1840 CrossRef CAS; (f) O. Sato, J. Tao and Y. Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS.
- (a) K. Awaga, T. Sekine, M. Ōkawa, W. Fujita, S. M. Holmes and G. S. Girolami, Chem. Phys. Lett., 1998, 293, 352–356 CrossRef CAS; (b) M. Mito, T. Kawae, K. Takeda, S. Takagi, Y. Matsushita, H. Deguchi, J. M. Rawson and F. Palacio, Polyhedron, 2001, 20, 1509–1512 CrossRef CAS; (c) V. Ksenofontov, G. Levchenko, S. Reiman, P. Gütlich, A. Bleuzen, V. Escax and M. Verdaguer, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 024415–244156 CrossRef; (d) E. Coronado, M. C. Giménez-López, G. Levchenko, F. M. Romero, V. García-Baonza, A. Milner and M. Paz-Pasternak, J. Am. Chem. Soc., 2005, 127, 4580–4581 CrossRef CAS; (e) W. Kaneko, M. Mito, S. Kitagawa and M. Ohba, Chem.–Eur. J., 2008, 14, 3481–3489 CrossRef CAS; (f) M. Ohba, W. Kaneko, S. Kitagawa, T. Maeda and M. Mito, J. Am. Chem. Soc., 2008, 130, 4475–4484 CrossRef CAS.
- (a) J. Larionova, S. A. Chavan, J. V. Yakhmi, A. G. Frøystein, J. Sletten, C. Sourisseau and O. Kahn, Inorg. Chem., 1997, 36, 6374–6381 CrossRef; (b) O. Kahn, J. Larionova and J. V. Yakhmi, Chem.–Eur. J., 1999, 5, 3443–3449 CrossRef CAS; (c) H. Miyasaka, N. Matsumoto, N. Re, E. Gallo and C. Floriani, Inorg. Chem., 1997, 36, 670–676 CrossRef CAS; (d) H. Miyasaka, H. Ieda, N. Matsumoto, N. Re, R. Crescenzi and C. Floriani, Inorg. Chem., 1998, 37, 255–263 CrossRef CAS; (e) N. Usuki, M. Ohba and H. Ōkawa, Bull. Chem. Soc. Jpn., 2002, 75, 1693–1698 CrossRef CAS.
- (a) D. Maspoch, D. Ruiz-Molina and J. Veciana, Chem. Soc. Rev., 2007, 36, 770–818 RSC; (b) C. J. Kepert, Chem. Commun., 2006, 695–700 RSC; (c) D. Maspoch, D. Ruiz-Molina, K. Wurst, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190–195 CrossRef CAS; (d) S. I. Ohkoshi, K. I. Arai, Y. Sato and K. Hashimoto, Nat. Mater., 2004, 3, 857–861 CrossRef CAS; (e) Z. Wang, B. Zhang, H. Fujiwara, H. Kobayashi and M. Kurmoo, Chem. Commun., 2004, 416–417 RSC; (f) M. Kurmoo, H. Kumagai, K. W. Chapman and C. J. Kepert, Chem. Commun., 2005, 3012–3014 RSC; (g) H. B. Cui, K. Takahashi, Y. Okano, H. Kobayashi, Z. Wang and A. Kobayashi, Angew. Chem., Int. Ed., 2005, 44, 6508–6512 CrossRef CAS; (h) N. Yanai, W. Kaneko, K. Yoneda, M. Ohba and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 3496–3497 CrossRef CAS; (i) W. Kaneko, M. Ohba and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 13706–13712 CrossRef CAS; (j) S. I. Ohkoshi, Y. Tsunobuchi, H. Takahashi, T. Hozumi, M. Shiro and K. Hashimoto, J. Am. Chem. Soc., 2007, 129, 3084–3085 CrossRef CAS; (k) Z. Wang, Y. Zhang, T. Liu, M. Kurmoo and S. Cao, Adv. Funct. Mater., 2007, 17, 1523–1536 CrossRef CAS; (l) J. Milon, M. C. Daniel, A. Kaiba, P. Guionneau, S. Brandés and J. P. Sutter, J. Am. Chem. Soc., 2007, 129, 13872–13878 CrossRef CAS; (m) B. Nowicka, M. Rams, K. Stadnicka and B. Sieklucka, Inorg. Chem., 2007, 46, 8123–8125 CrossRef CAS.
- E. Coronado and D. Gatteschi, J. Mater. Chem., 2006, 16, 2685–2515 RSC.
- (a) S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS; (b) N. R. Champness, Dalton Trans., 2006, 877–880 RSC; (c) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (d) J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS; (e) M. J. Rosseinsky, Microporous Mesoporous Mater., 2004, 73, 15–30 CrossRef CAS; (f) C. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS; (g) J. F. Eubank, V. C. Kravtsov and M. Eddaoudi, J. Am. Chem. Soc., 2007, 129, 5820–5821 CrossRef CAS; (h) G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- (a) K. Uemura, S. Kitagawa, M. Kondo, K. Fukui, R. Kitaura, H. C. Chang and T. Mizutani, Chem.–Eur. J., 2002, 8, 3586–3600 CrossRef CAS; (b) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, S. Horike and M. Takata, J. Am. Chem. Soc., 2004, 126, 14063–14070 CrossRef CAS.
- (a) T. Mallah, S. Thiebaut, M. Verdaguer and P. Veillet, Science, 1993, 262, 1554–1557 CrossRef CAS; (b) S. Ferlay, T. Mallah, R. Ouahés, P. Veillet and M. Verdaguer, Nature, 1995, 378, 701–703 CrossRef CAS; (c) M. Verdaguer, A. Bleuzen, V. Marvaud, J. Vaissermann, M. Seuleiman, C. Desplanches, A. Scuiller, C. Train, R. Garde, G. Gelly, C. Lomenech, I. Rosenman, P. Veillet, C. Cartier and F. Villain, Coord. Chem. Rev., 1999, 190–192, 1023–1047 CrossRef CAS.
- (a) S. S. Kaye, H. J. Choi and J. R. Long, J. Am. Chem. Soc., 2008, 130, 16921–16925 CrossRef CAS; (b) S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506–6507 CrossRef; (c) K. W. Chapman, P. D. Southon, C. L. Weeks and C. J. Kepert, Chem. Commun., 2005, 3322–3324 RSC.
- (a) M. Ohba and H. Ōkawa, Coord. Chem. Rev., 2000, 198, 313–328 CrossRef CAS; (b) H. Ōkawa and M. Ohba, Bull. Chem. Soc. Jpn., 2002, 75, 1191–1203 CrossRef; (c) M. Ohba, N. Maruono, H. Ōkawa, T. Enoki and J. M. Latour, J. Am. Chem. Soc., 1994, 116, 11566–11567 CrossRef CAS; (d) M. Ohba, H. Ōkawa, N. Fukita and Y. Hashimoto, J. Am. Chem. Soc., 1997, 119, 1011–1019 CrossRef CAS; (e) M. Ohba, N. Usuki, N. Fukita and H. Ōkawa, Angew. Chem., Int. Ed., 1999, 38, 1795–1798 CrossRef CAS; (f) K. Inoue, H. Imai, P. S. Ghalsasi, K. Kikuchi, M. Ohba, H. Ōkawa and J. V. Yakhmi, Angew. Chem., Int. Ed., 2001, 40, 4242–4245 CrossRef CAS; (g) K. Inoue, K. Kikuchi, M. Ohba and H. Ōkawa, Angew. Chem., Int. Ed., 2003, 42, 4810–4813 CrossRef CAS; (h) T. Shiga, H. Ōkawa, S. Kitagawa and M. Ohba, J. Am. Chem. Soc., 2006, 128, 16426–16427 CrossRef CAS.
- M. Ohba, K. Yoneda, G. Agustí, M. C. Muñoz, A. B. Gaspar, J. A. Real, M. Yamasaki, H. Ando, Y. Nakao, S. Sakaki and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4767–4771 CrossRef CAS.
- (a) Y. Yoshida, K. Inoue and M. Kurmoo, Chem. Lett., 2008, 37, 586–587 CrossRef CAS; (b) Y. Yoshida, K. Inoue and M. Kurmoo, Chem. Lett., 2008, 37, 504–505 CrossRef CAS; (c) Y. Yoshida, K. Inoue and M. Kurmoo, Inorg. Chem., 2009, 48, 267–276 CrossRef CAS.
- (a) “Spin Crossover in Transition Metal Compounds” (Eds: P. Gütlich, H. A. Goodwin), Top. Curr. Chem., 2004, 233–235 Search PubMed; (b) J. A. Real, A. B. Gaspar and M. Carmen Muñoz, Dalton Trans., 2005, 2062–2079 RSC; (c) H. Spiering, T. Kohlhaas, H. Romstedt, A. Hauser, C. Bruns-Yilmaz, J. Kusz and P. Gütlich, Coord. Chem. Rev., 1999, 190–192, 629–647 CrossRef CAS; (d) S. Hayami, R. Kawajiri, G. Juhász, T. Kawahara, K. Hashiguchi, O. Sato, K. Inoue and Y. Maeda, Bull. Chem. Soc. Jpn., 2003, 76, 1207–1213 CrossRef CAS; (e) P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef; (f) P. Gütlich, Y. Garcia and T. Woike, Coord. Chem. Rev., 2001, 219–221, 839–879 CrossRef CAS; (g) J. A. Real, A. B. Gaspar, V. Niel and M. C. Muñoz, Coord. Chem. Rev., 2003, 236, 121–141 CrossRef CAS; (h) A. Hauser, J. Jeftic, H. Romstedt, R. Hinek and H. Spiering, Coord. Chem. Rev., 1999, 190–192, 471–491 CrossRef CAS.
- (a) O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS; (b) V. Niel, J. M. Martinez-Agudo, M. C. Muñoz, A. B. Gaspar and J. A. Real, Inorg. Chem., 2001, 40, 3838–3839 CrossRef CAS.
- (a) J. A. Real, E. Andrés, M. C. Muñoz, M. Julve, T. Granier, A. Bousseksou and F. Varret, Science, 1995, 268, 265–267 CrossRef CAS; (b) G. J. Halder, K. W. Chapman, S. M. Neville, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2008, 130, 17552–17562 CrossRef CAS; (c) G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS; (d) S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, P. D. Southon, J. D. Cashion, J.-F. Letard, B. Moubaraki, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2008, 130, 2869–2876 CrossRef CAS; (e) V. Niel, A. L. Thompson, M. C. Muñoz, A. Galet, A. E. Goeta and J. A. Real, Angew. Chem., Int. Ed., 2003, 42, 3760–3763 CrossRef CAS.
- (a) S. Cobo, D. Ostrovskii, S. Bonhommeau, L. Vendier, G. Molnár, L. Salmon, K. Tanaka and A. Bousseksou, J. Am. Chem. Soc., 2008, 130, 9019–9024 CrossRef CAS; (b) G. Molnár, S. Cobo, J. A. Real, F. Carcenac, E. Daran, C. Vieu and A. Bousseksou, Adv. Mater., 2007, 19, 2163–2167 CrossRef CAS; (c) S. Bonhommeau, G. Molnár, A. Galet, A. Zwick, J. A. Real, J. J. McGarvey and A. Bousseksou, Angew. Chem., Int. Ed., 2005, 44, 4069–4073 CrossRef CAS; (d) T. Tayagaki, A. Galet, G. Molnár, M. C. Muñoz, A. Zwick, K. Tanaka, J. A. Real and A. Bousseksou, J. Phys. Chem. B, 2005, 109, 14859–14867 CrossRef CAS; (e) I. Boldog, A. B. Gaspar, V. Martínez, P. Pardo-Ibáñez, V. Ksenofontov, A. Bhattacharjee, P. Gütlich and J. A. Real, Angew. Chem., Int. Ed., 2008, 47, 6433–6437 CrossRef CAS; (f) F. Volatron, L. Catala, E. Riviére, A. Gloter, O. Stéphan and T. Mallah, Inorg. Chem., 2008, 47, 6584–6586 CrossRef CAS.
Footnote |
| † CCDC reference numbers 645084, 746507 and 746508. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b906442g |
| This journal is © The Royal Society of Chemistry 2010 |