DOI:
10.1039/B814040E
(Paper)
J. Mater. Chem., 2009,
19, 97-103
Highly fluorescent photochromic diarylethene with an excellent fatigue property†
Received
12th August 2008
, Accepted 22nd October 2008
First published on 17th November 2008
Abstract
A series of diarylethenes and their disulfonyl derivatives such as 1,2-bis(2-heptyl-1-benzothiophene-3-yl)perfluorocyclopentene (HBTF6), 1,2-bis(6-acetyl-2-heptyl-1-benzothiophene-3-yl)perfluorocyclopentene (DAHBTF6), 1,2-bis(2-heptyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentene (HBTFO4), and 1,2-bis(6-acetyl-2-heptyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentene (DAHBTFO4), have been prepared for the examination of a substituent and an oxidation effect on photochromic and photophysical properties. Steady state fluorescence studies indicated that the disulfonyl diarylethenes were highly fluorescent only in the closed form. The introduction of acetyl groups at the 6,6′ positions and heptyl groups at the 2,2′ positions in disulfonyl diarylethene gave rise to an increase in the fluorescence quantum yield and fluorescence lifetime. Fatigue properties of oxidized diarylethenes were improved by the introduction of acetyl groups, indicating that the photochromic properties of diarylethene are strongly affected by substituents. We also demonstrated the recording to and erasing of information from the materials by an alternating illumination with UV and visible light, and the accompanying strong fluorescence intensity changes provided a highly efficient information readout system, which may be applicable to erasable optical data-storage elements.
Introduction
Photochromic materials have received intense interest since their potential offers promise for photonic applications such as optical data storage, optical memory and switching devices (Scheme 1).1–7 In order to find application as an optical memory device, a photochromic material should have a good thermal stability and a high fatigue resistance.8–15 The ability to predictably control these properties by varying the structure will be essential to develop photochromic materials for various applications. For example, among the photochromic materials, diarylethenes containing benzothiophene derivatives such as 1,2-bis(2-methyl-1-benzothiophene-3-yl)perfluorocyclopentene (BTF6) particularly exhibit a good thermal stability and a high fatigue resistance.15 These properties can be improved further by varying the main body structure and/or substituents in the BTF6. In addition, the oxidation of sulfide to sulfone in the diarylethene varies the fatigue resistance and photophysical properties.16–19 Thus, those properties of diarylethenes required for a specific application can be optimized by controlling substituents and/or the oxidation state of the diarylethenes.
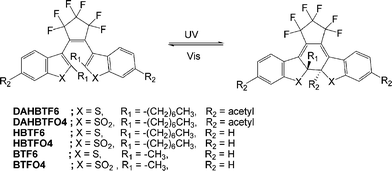 |
| | Scheme 1 Photochromic reactions of BTF6 and BTFO4 derivatives. | |
The photochromic phenomena can be observed by detecting either absorption or fluorescence changes between open- and closed-ring states of the diarylethene. We have focused our efforts on the fluorescence detection method because of its high sensitivity that may allow minimal damage to data while reading information.20–30 To accomplish the photocontrol and direct observation of those two isomerization states, ultimately at the single molecular level, it is essential to develop a photochromic molecule with a high fluorescence quantum yield and high photostability. Recently, we reported 1,2-bis(2-methyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentene (BTFO4), which is highly fluorescent in its closed-ring isomer state.16–18 Unfortunately, the low fatigue resistance and poor solubility of BTFO4 in common organic solvents limit its applications.17–19
Thus, we report herein the synthesis and characterization of a new series of oxidized diarylethene molecules including HBTFO4 and DAHBTFO4. In particular, DAHBTFO4 shows a high fluorescence quantum yield in the closed-ring isomer state with high fatigue resistance, good thermal stability and a good solubility in organic solvent which makes it suitable for photonic applications.
Results and discussion
Synthesis
HBTF6, DAHBTF6 and HBTFO4 were prepared from benzothiophene and 1-bromoheptane using the method previously described.17,31,32 DAHBTFO4 was synthesized from HBTF6 according to Scheme 2. The structures of the diarylethenes were confirmed by HRMS and 1H-NMR studies. Especially, single crystals of open ring isomers of HBTF6 (o-HBTF6) and HBTFO4 (o-HBTFO4) were obtained from CH2Cl2/methanol solution. Fig. 1 shows the ORTEP drawings of the molecular structure of HBTF6 in its photoactive antiparallel (A-P) conformation33 and HBTFO4 in the photoinactive parallel (P) conformation.34
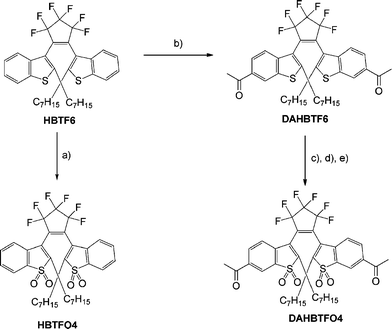 |
| | Scheme 2 Synthesis of diarylethenes; reaction conditions: (a) m-CPBA, CH2Cl2, 90%; (b) AlCl3, acetyl chloride, nitrobenzene, 82%; (c) ethylene glycol, p-TosH, benzene, 96%; (d) m-CPBA, CH2Cl2, 93%; (e) HCl, THF, 80%. | |
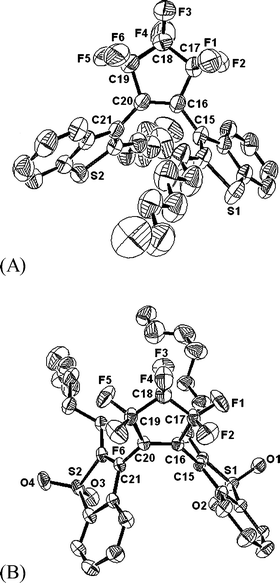 |
| | Fig. 1 ORTEP drawings of the open-ring isomer of (A) HBTF6 and (B) HBTFO4 with 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 16500 M−1cm−1 compared with BTFO4. In addition, the introduction of acetyl groups at the 6 and 6′ positions (DAHBTFO4) resulted in the further red shift of the absorption peak to 430 nm and an increase in molar absorption extinction coefficient to 24700 M−1cm−1. Upon illumination with 312 nm light, most of the open-ring isomer of DAHBTFO4 (o-DAHBTFO4) was converted to the closed-ring isomer (c-DAHBTFO4) according to 1H-NMR analysis showing an excellent photoconversion efficiency (>99%) which is far better than that of HBTFO4 (85%). The photochromic behaviors of HBTF6, HBTFO4, DAHBTF6, and DAHBTFO4 are similar to those of the previously reported compounds such as BTF6, BTFO4, 1,2-bis(6-acetyl-2-methyl-1-benzothiophene-3-yl)perfluorocyclopentene (DABTF6) and 1,2-bis(6-acetyl-2-methyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentene (DABTFO4).17 The photochromic properties, antiparallel (A-P) conformer ratios, cyclization quantum yields and ring opening quantum yields, of diarylethenes are summarized in Table 1. As shown in the table, the introduction of heptyl groups at the 2 and 2′ positions improved the photoactive antiparallel (A-P) conformer ratio and cyclization quantum yield compared with BTF6.35
000 to 16500 M−1cm−1 compared with BTFO4. In addition, the introduction of acetyl groups at the 6 and 6′ positions (DAHBTFO4) resulted in the further red shift of the absorption peak to 430 nm and an increase in molar absorption extinction coefficient to 24700 M−1cm−1. Upon illumination with 312 nm light, most of the open-ring isomer of DAHBTFO4 (o-DAHBTFO4) was converted to the closed-ring isomer (c-DAHBTFO4) according to 1H-NMR analysis showing an excellent photoconversion efficiency (>99%) which is far better than that of HBTFO4 (85%). The photochromic behaviors of HBTF6, HBTFO4, DAHBTF6, and DAHBTFO4 are similar to those of the previously reported compounds such as BTF6, BTFO4, 1,2-bis(6-acetyl-2-methyl-1-benzothiophene-3-yl)perfluorocyclopentene (DABTF6) and 1,2-bis(6-acetyl-2-methyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentene (DABTFO4).17 The photochromic properties, antiparallel (A-P) conformer ratios, cyclization quantum yields and ring opening quantum yields, of diarylethenes are summarized in Table 1. As shown in the table, the introduction of heptyl groups at the 2 and 2′ positions improved the photoactive antiparallel (A-P) conformer ratio and cyclization quantum yield compared with BTF6.35
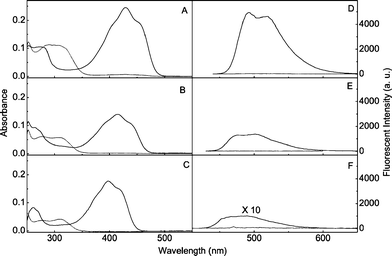 |
| | Fig. 2 (A, B and C) Electronic ground state absorption and (D, E and F) steady state fluorescence spectra of DAHBTFO4 (A, D), HBTFO4 (B, E) and BTFO4 (C, F) for open-ring (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and photostationary state (—) isomers in ethyl acetate (1.0 × 10−5 M) at room temperature. ) and photostationary state (—) isomers in ethyl acetate (1.0 × 10−5 M) at room temperature. | |
Table 1 Photochromic properties of diarylethenes
| |
AP:Pa |
Φ
o→c
b
,
c
|
Φ
c→o
b
,
d
|
Conversion at PSS (%)e |
λ
max (nm)/ε (103 M−1 cm−1) |
| Open ring isomer |
Closed ring isomer |
|
Antiparallel (AP) to parallel (P) ratio. Observed in 1H NMR spectra in CDCl3 (5.0 × 10−3 M).
BTF6 was used as reference 43.
Cyclization quantum yield, measured at 312 nm in ethyl acetate.
Ring opening quantum yield in ethyl acetate, measured at λmax of closed-ring isomer.
Conversion at photostationary state (PSS) was calculated from the 1H NMR spectra in CDCl3 (5.0 × 10−3 M). For all compounds the conversion of the ring opening was 1.
Taken from reference 43.
|
| BTF6f |
65:35 |
0.31 |
0.28 |
43 |
258/16 |
276/14.0 |
| 290/6.2 |
352/12.0 |
| 299/6.8 |
523/10.0 |
| DABTF6 |
65:35 |
0.46 |
0.15 |
73 |
286/28.6 |
276/16.2 |
| 316/12.0 |
368/16.0 |
| 324/12.8 |
552/10.8 |
| HBTF6 |
77:23 |
0.34 |
0.33 |
65 |
252/18.6 |
288/11.5 |
| 290/6.5 |
256/7.8 |
| 300/6.7 |
544/6.7 |
| DAHBTF6 |
75:25 |
0.49 |
0.19 |
99 |
286/33.1 |
278/19.0 |
| 314/13.2 |
274/12.5 |
| 324/12.7 |
572/8.9 |
| BTFO4 |
50:50 |
0.22 |
0.061 |
80 |
276/3.7 |
262/9.1 |
| 308/4.1 |
398/21.0 |
| DABTFO4 |
50:50 |
0.40 |
0.055 |
99 |
302/11.1 |
274/10.6 |
| — |
412/27.9 |
| HBTFO4 |
64:36 |
0.23 |
0.38 |
85 |
278/6.1 |
264/10.0 |
| 310/5.7 |
414/16.5 |
| DAHBTFO4 |
60:40 |
0.45 |
0.21 |
99 |
290/11.3 |
274/10.5 |
| 306/11.0 |
430/24.7 |
Fluorescence spectra, fluorescence quantum yields, and fluorescence lifetimes
Upon exposure to UV light, o-HBTFO4 and o-DAHBTFO4 were photocyclized to c-HBTFO4 and c-DAHBTFO4, respectively, along with a significant increase in the fluorescence intensities. Steady-state fluorescence spectra of open-ring (dashed line) and closed-ring (solid line) isomers of DAHBTFO4 and HBTFO4 with 400 nm photo-excitation in ethyl acetate (1.0 × 10−5 M) are shown in Fig. 2D and 2E, respectively. For comparison, the fluorescence spectra of o-BTFO4 (dashed line) and c-BTFO4 (solid line) are also shown in Fig. 2F. Comparison of the fluorescence spectra of those diarylethenes revealed substituent effects similar to the absorption spectra. The fluorescence bands were red-shifted to 506 nm and 502 nm by introduction of acetyl groups (c-DABTFO4) and heptyl groups (c-HBTFO4) (Fig. 2E), respectively, compared with c-BTFO4 (492 nm).17 In the case of DAHBTFO4, the presence of both substituents resulted in further red-shift to 515 nm (Fig. 2D), as expected, due to the combined effect of both substituents. Fig. 2 also indicates that the introduction of acetyl groups at the 6 and 6′ positions and a long alkyl group instead of methyl at the 2 and 2′ positions of the BTFO4 significantly enhances the fluorescence intensity. Fig. 3 shows photographs exhibiting the emission of the solutions containing DAHBTFO4 and BTFO4 in ethyl acetate (1.0 × 10−4 M). The colorless solutions containing o-DAHBTFO4 and o-BTFO4 (Fig. 3A and 3C) were changed to closed-ring isomers upon exposure to UV light, which emit strong green light upon 400 nm photoexcitation (Fig. 3B and 3D). The photographs clearly show that the fluorescence emission of closed-ring isomer of DAHBTFO4 is much stronger than that of BTFO4.
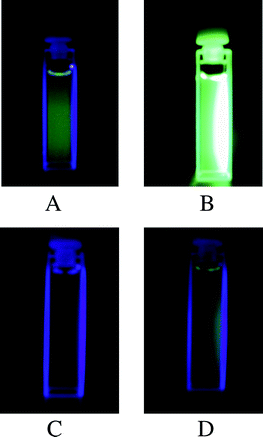 |
| | Fig. 3 Photographs of solutions containing, in ethyl acetate (1 × 10−4 M) under 400 nm light, DAHBTFO4 (A) as the open ring isomer before UV irradiation and (B) as the photostationary state after UV irradiation, and BTFO4 (C) as the open ring isomer before UV irradiation and (D) as the photostationary state after UV irradiation. | |
To clarify the substitution effect on the fluorescence, the fluorescence quantum yields were determined by using 3-aminofluoranthene (0.53 in cyclohexane) as the reference upon 400 nm photo-excitation. The fluorescence quantum yields of the c-HBTFO4 and c-DAHBTFO4 are given in Table 2. For comparison, Table 2 also contains the quantum yields of c-BTFO4 and c-DABTFO4 reported previously. Interestingly, the fluorescence quantum yield of c-DAHBTFO4 was measured to be 0.52, which is the best value among the diarylethenes reported thus far in their closed form. The fluorescence quantum yields of c-DABTFO4 (0.036) and c-HBTFO4 (0.19) were also higher than that of BTFO4 (0.01).17 From those data, it was found that the displacement of the methyls at the 2,2′ positions of the benzothiophene subunit by heptyl groups (c-HBTFO4) gave rise to an increase in the quantum yield by about 19 times, whereas the introduction of acetyl substituents at the 6,6′ positions (c-DABTFO4) produced about 4 times increase compared with BTFO4. The presence of both substituents (c-DAHBTFO4) resulted in a significant increase in the quantum yield by about 52 times due to the combined effect of the two substituents. To explain the difference in the fluorescence quantum yields, the excited state lifetimes of those diarylethenes were measured.
Table 2 Fluorescence properties of diarylethenes
| Compound |
λ
em (nm)a |
Φ
F
b
|
τ (ns) |
|
λ
max of emission band following photo-excitation at 400 nm.
Determined by using 3-aminofluoranthene (0.53 in cyclohexane) as the reference.
|
| c-DAHBTFO4 |
492, 520 |
0.52 |
1.7 |
| c-HBTFO4 |
476, 502 |
0.19 |
0.73 |
| c-DABTFO4 |
506 |
0.036 |
0.17 |
| c-BTFO4 |
492 |
0.01 |
0.06 |
To elucidate the origin of the high fluorescence quantum yield of c-DAHBTFO4, we have performed time-correlated single photon counting (TCSPC) fluorescence measurements in ethyl acetate solution. Fig. 4 shows the time-resolved fluorescence decay profiles of c-BTFO4, c-DABTFO4, c-HBTFO4, and c-DAHBTFO4 in ethyl acetate at room temperature. The excitation wavelength was 400 nm and monitored at 500 nm. The fluorescence decay curves exhibited single-exponential decays and the fitting parameters obtained from the fluorescence decay curves are listed in Table 2. These results clearly indicate that the fluorescence lifetimes are strongly affected by the substituents. The fluorescence lifetimes of c-HBTFO4 (0.73 ns) and c-DABTFO4 (0.17 ns) were much longer than that of c-BTFO4 (0.06 ns). These results imply that the long alkyl chains at the 2,2′ positions substantially affects the decay rate of the fluorescence compared with acetyl substituents at the 6,6′ positions. In addition, we can also expect that the introduction of the two substituents at both positions will even further increase the fluorescence lifetime, as observed with c-DAHBTFO4 (1.7 ns). As shown in Fig. 4 and Table 2, the fluorescence lifetimes have a tendency similar to fluorescence quantum yields, which indicates that the significant increase in fluorescence quantum yield originates from the increase in the fluorescence lifetime.
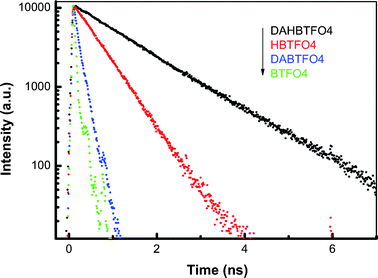 |
| | Fig. 4 The time-resolved fluorescence decay profiles of BTFO4, DABTFO4, HBTFO4, and DAHBTFO4. | |
Fluorescence modulation
Upon exposure to UV light, the open-ring isomers of diarylethenes with sulfone groups were photocyclized to their closed-ring isomers with high cyclization yields accompanied by a significant increase in the fluorescence intensity. Since the fluorescence quantum yields of o-DAHBTFO4 and o-HBTFO4 are very low, unlike to their closed-ring isomers, c-DAHBTFO4 and c-HBTFO4 (Fig. 2D and 2E), the modulation of the fluorescence intensity through photochromic conversion can be performed. Thus, we investigated the fluorescence intensity changes of HBTFO4 and DAHBTFO4 in ethyl acetate at room temperature using alternating UV and visible light illumination. Fig. 5 illustrates the fluorescence modulation of DAHBTFO4 (closed circles) and HBTFO4 (closed triangles) recorded at 520 nm (λex = 428 nm) and 502 nm (λex = 414 nm), respectively, as a function of time using alternating UV (solid line) and visible (dashed line) light irradiation. Upon UV irradiation, the fluorescence intensities were increased whereas the fluorescence intensities were decreased upon visible light illumination. The highest photo-induced fluorescence modulation was observed from DAHBTFO4 compared with those from HBTFO4 and previously reported BTFO4, as expected. The fluorescence contrast between the closed-ring and open-ring isomers (max/min ratio) of DAHBTFO4 was 140, whereas those of HBTFO4 and BTFO4 were 53 and 3, respectively. Such a high fluorescence contrast must have been the result of the high fluorescence quantum yield of c-DAHBTFO4. While c-DAHBTFO4 maintained its fluorescence intensity during the modulation for 10 times, the fluorescence intensity of c-HBTFO4 slowly decreased. This result is in accord with the relatively low fatigue property of HBTFO4 that will be discussed later. We have not observed any fluorescence intensity decay for DAHBTFO4 while reading the signal with 428 nm light for 10 cycles, indicating that DAHBTFO4 can be applied as an active medium for a highly efficient optical readout system in the optical memory or all-optical switching device.
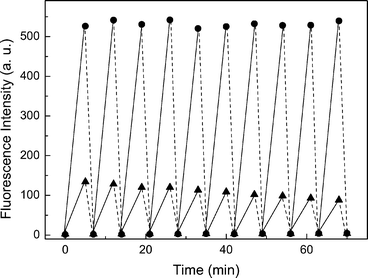 |
| | Fig. 5 Modulation of fluorescence signals of DAHBTFO4 (λex = 428 nm, λem = 520 nm, ●) and HBTFO4 (λex = 414 nm, λex = 502 nm, ▲) in ethyl acetate (1.0 × 10−5 M) upon alternating 312 nm (—) and visible () light illumination as a function of time. | |
To demonstrate the potential of the material for a high density optical memory device application, a film of DAHBTFO4 in poly(methyl methacrylate) (PMMA) matrix (5 wt%) was prepared. Fig. 6 shows the fluorescence images of a DAHBTFO4 film following 488 nm photoexcitation with a CARL ZEISS confocal microscope. Initially a letter A mask pattern was placed on the film which was then irradiated with 315 nm UV light, which gave rise to the formation of the closed-ring isomer along the mask. Thus, we were able to observe the fluorescence image of the letter A (Fig. 6 left). The fluorescence signal was erased upon the irradiation of visible light on the film because of the ring-opening of the closed-ring isomer (Fig. 6 centre). Then an E mask pattern was placed on the film which was then irradiated with 315 nm UV light. We could again observe the fluorescence image of the letter E. Therefore, we clearly demonstrated that the alternating illumination with UV and visible light on the film containing DAHBTFO4 can result in recording and erasing information, and the highly efficient information readout can be carried out using the observation of the fluorescence intensity changes.
 |
| | Fig. 6 Photo-rewritable fluorescence imaging on 5 wt% in PMMA film by using UV and visible light. The real size of the photomask is about 1 mm × 1.2 mm. | |
Photochromic properties
We investigated the fatigue properties of various diarylethenes in ethyl acetate (1.0 × 10−5 M) at room temperature by applying UV light irradiation for 10 h.17–19,26Fig. 7 illustrates the absorbance changes of diarylethenes (BTF6, open square; BTFO4, closed square; HBTF6, open triangle; HBTFO4, closed triangle; DAHBTF6, open circle, DAHBTFO4, closed circle) at the absorption maximum of their closed-ring isomer as a function of UV illumination time. The absorbance decrease could be attributed mainly to the by-product formation from the closed-ring isomer.36–39 At the end of each experiment, absorption spectra of those solutions were obtained after visible light irradiation to verify that the by-products did not have absorption peaks near to λmax of their closed-ring isomer. The important features of these experiments are that both the oxidation of sulfide to sulfone and the substitution of long alkyl chains at the 2 and 2′ positions of the benzothiophene subunit result in the decrease of the photostability of the diarylethenes. However, the substitution of acetyl groups at the 6 and 6′ positions of benzothiophene subunits leads to a significant improvement in the photostability.17 As seen in Fig. 7, the fatigue resistance of DAHBTFO4 (4600 min) is about 3 times better than that of BTF6 (1400 min) which has been regarded as a good fatigue resistance material.
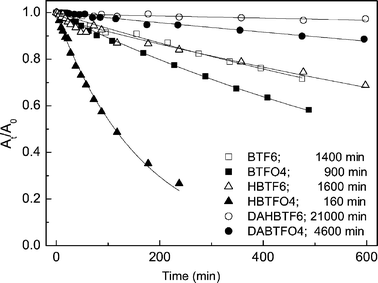 |
| | Fig. 7 The absorbance changes of BTF6 (□), BTFO4 (■), HBTF6 (△), HBTFO4 (▲), DAHBTF6 (○) and DAHBTFO4 (●) at the absorption maximum of the closed-ring isomer as a function of UV illumination time in ethyl acetate (1 × 10−5 M). The data are fitted with exponential decay (solid line) and the fitting data (photoreaction time) represent the time to reach 37% of the initial absorbance following UV illumination. | |
We also investigated the thermal stability of the closed-ring isomers of BTF6, HBTF6, HBTFO4, DAHBTF6 and DAHBTFO4 in mesitylene (2.0 × 10−5 M) at 80 °C under dark conditions and found that the oxidation of sulfide to sulfone and the introduction of acetyl groups at the 6 and 6′ positions lead to a big improvement in the thermal stability compared with BTF6 and HBTF6.40,41
Conclusion
We have prepared a new highly fatigue-resistant and thermally stable diarylethene, DAHBTFO4, by introduction of heptyl and acetyl groups at the 2,2′ and 6,6′ positions of BTFO4, respectively. Photophysical and photochromic studies, including the fluorescence quantum yield, fatigue property and thermal stability, have shown that those properties of DAHBTFO4 were significantly improved compared with the previously reported BTFO4 and BTF6. In addition, we have demonstrated that the alternating photostimulation by UV and visible light of a film of DAHBTFO4 can lead to fluorescence switching. These results reveal the potential of DAHBTFO4 as a key material in various optical applications such as optical memory, all-optical switches and other optics parts based on a reversible photochromic conversion.
Experimental section
General methods
Octafluorocyclopentene was purchased from TCI. All other reagents and spectroscopic grade solvents were purchased from Aldrich. Melting points were determined with a Laboratory Devices Mel-Temp 3.0 melting point apparatus. The 1H and 13C NMR spectra were obtained using a Jeol JNM-AL300 spectrometer at 300 MHz and 75 MHz in CDCl3, respectively, with tetramethylsilane as the internal reference. HRMS spectra were obtained with a Jeol JMS-700 spectrometer. Absorption spectra were recorded on a Shimadzu UV-3100 spectrophotometer in spectroscopy grade ethyl acetate. Fluorescence spectra were collected in spectroscopy grade ethyl acetate on a Fluoro Max-2 spectrophotometer equipped with a 150 W ozone-free xenon lamp. UV and visible irradiations were performed with standard lamps used for visualizing TLC plates (VL6L; 312 nm, 8 mW cm−2) and a 100 W tungsten lamp and the samples were placed in a glass chamber maintained at room temperature. The quantum yields were measured using a 500 W Xe lamp (Newport 74000) combined with a monochromator (Newport 66921). Then, photochromic reaction quantum yields were determined according to the method described in references 42 and 43. For confocal laser scanning microscope experiments, a cover glass treated with “piranha” solution (4:1 mixture of concentrated H2SO4 and 30% H2O2) was used. The wavelength region for excitation and probing was 488 nm and 505–530 nm, respectively. The fluorescence micrographs were taken using an LSM 510 META (CARL ZEISS). The excitation wavelength was 442 nm from a He–Cd laser and an Avalanche photodiode (Perkin Elmer, SPCM-AQR-13-FC) was used for detecting fluorescence intensity. 1H-NMR (5 × 10−3 M, CDCl3) spectroscopy of closed-ring isomers and conversion were determined after irradiation at 312 nm using a UV lamp until the photostationary state was reached. X-Ray crystallographic analysis of single crystals was performed with a Bruker Smart Apex II X-ray diffractometer with Mokα radiation and a graphite monochromator. Crystal cell constants were calculated by global refinement. The structure was solved by direct method with SHELXS8644 and refined by full least squares on F2 with SHELX97.45
Synthesis
1,2-Bis(2-heptyl-1-benzothiophene-3-yl)perfluorocyclopentene (HBTF6).
3-Bromo-2-heptylbenzothiophene (1.0 g, 3.2 mmol) was dissolved in THF (12 mL). n-BuLi (2.5 M in hexane, 1.3 mL, 3.2 mmol) was added slowly with stirring at −78 °C. The reaction mixture was stirred for 30 min and then octafluorocyclopentene (0.22 mL, 1.6 mmol) was added rapidly at the same temperature. After stirring for 1 h, the mixture was warmed to room temperature and quenched with H2O. The mixture was extracted with CH2Cl2 (2 × 25 mL), dried over MgSO4, filtered and the solvent was removed. The residue was purified by chromatography on silica gel to give HBTF6 (0.33 g) as a white solid with 33% yield. Mp: 64–65 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.73–7.69 (m, p-4H), 7.62–7.55 (m, ap-4H), 7.40–7.28 (m, ap-4H), 7.18–7.15 (m, p-4H), 2.90–2.81 (m, p-2H), 2.70–2.55 (m, p-2H and ap-2H), 2.30–2.17 (m, ap-2H), 1.69–1.67 (m, p-2H), 1.30–0.90 (m, p-18H and p-20H), 0.87 (t, p-6H and ap-6H, J = 7.2 Hz). HRMS: m/z: calcd for C35H38F6S2: 636.2319; found: 636.2338.
1,2-Bis(6-acetyl-2-heptyl-1-benzothiophene-3-yl)perfluorocyclopentene (DAHBTF6).
To a nitrobenzene solution (11 mL) of HBTF6 (0.62 g, 0.97 mmol) and acetyl chloride (0.30 g, 4.87 mmol) was added AlCl3 (0.16 g, 4.87 mmol) at room temperature. The solution was stirred for 1 h at room temperature. After completion of the reaction, 1 N HCl (5 mL) was added. The mixture was extracted with CH2Cl2 (2 × 25 mL), dried over MgSO4, filtered and the solvents were removed. The residue was purified by chromatography on silica gel to give solid DAHBTF6 (0.58 g) with 82% yield. Mp: 101–103 °C. 1H NMR (CDCl3, 300 MHz): δ = 8.35 (d, ap-2H. J = 1.5 Hz), 8.23 (d, p-2H, J = 1.5 Hz), 7.97 (dd, ap-2H, J1 = 8.4 Hz, J2 = 1.5 Hz), 7.77–7.71 (m, ap-2H and p-2H), 7.56 (d, ap-2H, J = 8.4 Hz), 2.93–2.83 (m, p-2H), 2.75–2.59 (m, ap-2H and p-2H), 2.67 (s, ap-6H), 2.60 (s, p-6H), 2.31–2.22 (m, ap-2H), 1.88–1.64 (m, p-4H), 1.39–0.91 (m, ap-20H and p-16H), 0.833 (t, ap-6H and p-6H, J = 7.2 Hz). HRMS: m/z: calcd for C39H42F6O2S2: 720.2530; found: 720.2483.
1,2-Bis(6-(2,5-dioxolanyl-1-methyl)-2-heptyl-1-benzothiophene-3-yl)perfluorocyclopentene (DPHBTF6).
A solution of compound DAHBTF6 (1.2 g, 1.6 mmol), ethylene glycol (2 mL, 32 mmol) and p-toluenesulfonic acid monohydrate (61 mg, 0.32 mmol) in benzene (300 mL) was refluxed for 20 h with a Dean–Stark condenser. After completion of reaction, H2O (50 mL) was added, extracted with CH2Cl2 (2 × 50 mL) and washed with aqueous Na2CO3 (2 × 50 mL). The organic layer was dried with MgSO4, filtered and the solvent was removed. The residue was purified by chromatography on silica gel to give DPHBTF6 (1.2 g) as an oil of with 96% yield. 1H NMR (CDCl3, 300 MHz): δ 7.86 (s, ap-2H), 7.74 (s, p-2H), 7.66 (d, ap-2H, J = 8.4 Hz), 7.54–7.49 (m, p-2H and ap-2H), 7.27 (d, p-2H, J = 9.0 Hz), 4.08–4.00 (m, ap-4H and p-4H), 3.80–3.66 (m, ap-4H and p-4H), 3.14–3.09 (m, p-2H), 2.91–2.81 (m, p-2H), 2.72–2.59 (m, ap-2H), 2.39–2.23 (m, ap-2H), 1.70 (s, ap-6H), 1.60 (s, p-6H), 1.41–0.98 (m, ap-20H and p-20H), 0.91–0.85 (m, ap-6H and p-6H). HRMS: m/z: calcd for C43H50F6O4S2: 808.3055; found 808.3109.
1,2-Bis(6-(2,5-dioxolanyl-1-methyl)-2-heptyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentene (DPHBTFO4).
A mixture of compound DPHBTF6 (0.57 g, 0.70 mmol) and 3-chloroperbenzoic acid (77%, 1.2 g, 7.0 mmol) in CH2Cl2 (20 mL) was stirred for 24 h at room temperature. The solution was washed with a saturated solution of Na2SO4 (50 mL × 2) and NaHCO3 (50 mL × 2). The organic layer was separated and dried over MgSO4 and concentrated in vacuo. Flash chromatography (silica gel) yielded DPHBTFO4 (0.57 g) in 93% yield. Mp: 55–57 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.90 (s, ap-2H), 7.81 (s, p-2H), 7.73 (d, ap-2H, J = 8.1 Hz), 7.49 (d, p-2H, J = 7.8 Hz), 7.15 (d, ap-2H, J = 8.1 Hz), 7.07 (d, p-2H, J = 7.8 Hz), 4.08–4.03 (m, ap-4H and p-4H), 3.87–3.62 (m, ap-4H and p-4H), 2.82–2.77 (m, p-2H), 2.66–2.56 (m, p-2H), 2.48–2.38 (m, ap-2H), 2.34–2.24 (m, ap-2H), 1.64 (s, ap-6H), 1.57 (s, p-6H), 1.37–1.19 (m, ap-20H and p-20H), 0.90–0.86 (m, ap-6H and p-6H). HRMS: m/z: calcd for C43H50F6O8S2: 872.2851; found 872.2825.
1,2-Bis(6-acetyl-2-heptyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentene (DAHBTO4).
To a solution of DPHBTFO4 (0.57 g, 0.65 mmol) in THF (25 mL) was added 37% HCl (1 mL). The solution was stirred for 3 h at room temperature. After completion of the reaction, H2O (50 mL) was added, the mixture extracted with CH2Cl2 (2 × 50 mL) and washed with aqueous Na2CO3 (2 × 50 mL). The organic layer was dried with MgSO4, filtered and the solvents were removed. The residue was purified by chromatography on silica gel to give compound 7 (0.41 g) as a solid with 80% yield. Mp: 61–63 °C. 1H NMR (CDCl3, 300 MHz): δ = 8.30 (d, ap-2H, J = 1.8 Hz), 8.23–8.20 (m, p-2H and ap-2H), 8.02 (dd, p-2H, J1 = 7.8 Hz, J2 = 1.2 Hz), 7.32 (d, ap-2H, J = 7.8 Hz), 7.22 (d, p-2H, J = 7.8 Hz), 2.66 (s, ap-6H), 2.60 (s, p-6H), 2.49–2.39 (m, ap-4H), 2.32–2.22 (m, p-4H), 1.95–1.84 (m, p-2H), 1.80–1.65 (m, ap-4H), 1.39–0.99 (m, ap-16H and p-18H), 0.90–0.84 (m, ap-6H and p-6H). HRMS: m/z: calcd for C39H42F6O6S2: 784.2327; found 784.2341.
1,2-Bis(2-heptyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentene (HBTFO4).
HBTFO4 (0.49 g) was synthesized, from HBTF6 (0.50 g, 0.79 mmol) by the same method as compound DPHBTFO4, in 90% yield. Mp: 105–107 °C. 1H-NMR (CDCl3, 300 MHz): δ 7.77 (d, ap-2H, J = 7.8 Hz), 7.69–7.56 (m, p-2H and ap-4H), 7.48–7.27 (m, p-4H), 7.22 (d, ap-2H, J = 6.6 Hz), 7.17 (d, p-2H, J = 7.2 Hz), 2.72–7.55 (m, p-2H), 2.47–2.35 (m, p-2H and ap-2H), 2.28–2.02 (m, ap-2H), 1.99–1.82 (m, p-2H), 1.77–1.63 (m, p-2H and ap-2H), 1.36–1.03 (m, p-16H and ap-18H), 0.87 (t, p-6H and ap-6H, J = 7.2 Hz). HRMS: m/z: calcd for C35H38F6O4S2: 700.2116; found 700.2142.
Acknowledgements
This research was supported by the GRRC Project of Gyeonggi Provincial Government, Republic of Korea (S. I. Y., K. H. A.). We would like to thank Professor S. Lee at SSKU for X-ray analysis.
References
- A. E. J. Wilson, Phys. Technol., 1984, 15, 232 Search PubMed.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327 RSC.
- H. Tian and Y. Feng, J. Mater. Chem., 2008, 18, 1617 RSC.
- S. Pu, T. Yang, B. Yao, Y. Wang, M. Lei and J. Xu, Mater. Lett., 2007, 61, 855 CrossRef CAS.
- A. J. Kronemeijer, H. B. Akkerman, T. Kudernac, B. J. van Wees, B. L. Feringa, P. W. M. Blom and B. de Boer, Adv. Mater., 2008, 20, 1467 CrossRef CAS.
- Z.-X. Li, L.-Y. Liao, W. Sun, C.-H. Xu, C. Zhang, C.-J. Fang and C.-H. Yan, J. Phys. Chem. C, 2008, 112, 5190 CrossRef CAS.
- Z. Zhang, X. Liu, Z. Li, Z. Chen, F. Zhao, F. Zhang and C.-H. Tung, Adv. Funct. Mater., 2008, 18, 302 CrossRef CAS.
- Y. Liu, Q. Wang, Y. Liu and X.-Z. Yang, Chem. Phys. Lett., 2003, 373, 338 CrossRef CAS.
- Y. Nakayama, K. Hayashi and M. Irie, J. Org. Chem., 1990, 55, 2592 CrossRef CAS.
- Y. Nakayama, K. Hayashi and M. Irie, Bull. Chem. Soc. Jpn., 1991, 64, 789 CAS.
- K. Morimitsu, K. Shibata, S. Kobatake and M. Irie, J. Org. Chem., 2002, 67, 4574 CrossRef CAS.
- C. Zheng, S. Pu, J. Xu, M. Luo, D. Huang and L. Shen, Tetrahedron, 2007, 63, 5437 CrossRef CAS.
- H. Ikeda, A. Sakai, A. Kawabe, H. Namai and K. Mizuno, Tetrahedron Lett., 2008, 49, 4972 CrossRef CAS.
- D. Dulić, T. Kudernac, A. Pužys, B. L. Feringa and B. J. van Wees, Adv. Mater., 2007, 19, 2898 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS.
- Y.-C. Jeong, S. I. Yang, K.-H. Ahn and E. Kim, Chem. Commun., 2005, 2503 RSC.
- Y.-C. Jeong, S. I. Yang, E. Kim and K.-H. Ahn, Tetrahedron, 2006, 62, 5855 CrossRef CAS.
- Y.-C. Jeong, J. P. Han, Y. Kim, E. Kim, S. I. Yang and K.-H. Ahn, Tetrahedron, 2007, 63, 3173 CrossRef CAS.
- Y.-C. Jeong, D. G. Park, E. Kim, K.-H. Ahn and S. I. Yang, Chem. Commun., 2006, 1881 RSC.
- H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85 RSC.
- M. Bossi, V. Belov, S. Polyakova and S. W. Hell, Angew. Chem., Int. Ed., 2006, 45, 7462 CrossRef CAS.
- C. C. Corredor, Z.-L. Huang and K. D. Belfield, Adv. Mater., 2006, 18, 2910 CrossRef CAS.
- S.-J. Lim, J. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 45, 14542 CrossRef.
- G. Jiang, S. Wang, W. Yuan, L. Jiang, Y. Song, H. Tian and D. Zhu, Chem. Mater., 2006, 18, 235 CrossRef CAS.
- T. A. Golovkova, D. V. Kozlov and D. C. Neckers, J. Org. Chem., 2005, 70, 5545 CrossRef CAS.
- A. de Meijere, L. Zhao, V. N. Belov, M. Boss, M. Noltemeyer and S. W. Hell, Chem.–Eur. J., 2007, 13, 2503 CrossRef.
- J. H. Hurenkamp, J. J. D. de Jong, W. R. Browne, J. H. Van Esch and B. L. Feringa, Org. Biomol. Chem., 2008, 6, 1268 RSC.
- J. Finden, T. K. Kunz, N. R. Branda and M. O. Woif, Adv. Mater., 2008, 20, 1998 CrossRef CAS.
- S. Xiao, Y. Zou, M. Yu, T. Yi, Y. Zhou, F. Li and C. Huang, Chem. Commun., 2007, 4758 RSC.
- C. C. Corredor, Z.-L. Huang, K. D. Belfield, A. R. Morales and M. V. Bondar, Chem. Mater., 2007, 19, 5165 CrossRef CAS.
- M. Irie, O. Miyatake, K. Uchida and T. Eriguchi, J. Am. Chem. Soc., 1994, 116, 9894 CrossRef CAS.
- T. Yamaguchi and M. Irie, J. Photochem. Photobiol., A, 2006, 178, 162 CrossRef CAS.
- Crystal data for HBTF6: C35H38F6S2, MW = 636.77, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.8702(5) Å, b = 12.6588(6) Å, c = 14.6884(10) Å, α = 109.028(3)°, β = 99.410(3)°, γ = 111.268(2)°, V = 1687.08(16) Å3, T = 295 K, Z = 2, dcalc = 1.254 g cm−3, 33020 reflections collected, 8320 unique (Rint = 0.0242), no. parameters = 388, R1 = 0.0876 [4050 data with I > 2σ(I)], wR2 = 0.3405, GOF = 1.070, CCDC deposition number: 698528.
, a = 10.8702(5) Å, b = 12.6588(6) Å, c = 14.6884(10) Å, α = 109.028(3)°, β = 99.410(3)°, γ = 111.268(2)°, V = 1687.08(16) Å3, T = 295 K, Z = 2, dcalc = 1.254 g cm−3, 33020 reflections collected, 8320 unique (Rint = 0.0242), no. parameters = 388, R1 = 0.0876 [4050 data with I > 2σ(I)], wR2 = 0.3405, GOF = 1.070, CCDC deposition number: 698528.
- Crystal data for HBTFO4: C35H38F6O4S, MW = 700.77, triclinic, space group P, a = 10.7844(2) Å, b = 12.9936(2) Å, c = 14.1194(2) Å, α = 112.5730(10)°, β = 97.2540(10)°, γ = 98.2730(10)°, V = 1771.92(5) Å3, T = 295 K, Z = 2, dcalc = 1.313 g cm−3, 34163 reflections collected, 8744 unique (Rint = 0.0229), no. parameters = 424, R1 = 0.0780 [6295 data with I > 2σ(I)], wR2 = 0.2601, GOF = 1.059, CCDC deposition number: 698529.
- K. Uchida, E. Tsuchida, Y. Aoi, S. Nakamura and M. Irie, Chem. Lett., 1999, 63 CrossRef CAS.
- Y.-C. Jeong, E. Kim, K.-H. Ahn and S. I. Yang, Bull. Korean Chem. Soc., 2005, 26, 1675 CAS.
- A. Peters and N. R. Branda, Adv. Mater. Opt. Electron., 2000, 10, 245 CrossRef CAS.
- K. Higashiguchi, K. Matsuda, S. Kobatake, T. Yamada, T. Kawai and M. Irie, Bull. Chem. Soc. Jpn., 2000, 73, 2389 CrossRef CAS.
- M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747 RSC.
- S. Nakamura and M. Irie, J. Org. Chem., 1988, 53, 6136 CrossRef.
- S. Iwata, Y. Ishihara, C.-P. Qian and K. Tanaka, J. Org. Chem., 1992, 57, 3726 CrossRef CAS.
- H. Nakashma and M. Irie, Macromol. Chem. Phys., 1999, 200, 683 CrossRef.
- K. Matsuda and M. Irie, Tetrahedron Lett., 2000, 41, 2577 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
-
G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.