Green synthesis of a temperature sensitive hydrogel
Márcio
Temtem
a,
Teresa
Casimiro
a,
João F.
Mano
*bc and
Ana
Aguiar-Ricardo
*a
aREQUIMTE/CQFB, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516, Caparica, Portugal. E-mail: aar@dq.fct.unl.pt; Fax: +351 212 948 385; Tel: +351 21 2949648
b3B's Research Group—Biomaterials, Biodegradables and Biomimetics, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal. E-mail: jmano@dep.uminho.pt; Fax: +351 253604492; Tel: +351 253604497
cDepartment of Polymer Engineering, University of Minho, Campus de Azurém, 4810-058, Guimarães, Portugal
First published on 27th October 2006
Abstract
A thermoresponsive hydrogel, poly(N-isopropylacrylamide), PNIPAAm, with possible applications in drug delivery, tissue engineering and as smart membranes with tuned permeability, was synthesised in supercritical carbon dioxide. A strategy of solvent-free impregnation/coating of polymeric surfaces with PNIPAAm is also suggested, in order to further extend the applications of membranes or porous bulky systems. In this work, in situ synthesis of PNIPAAm within a chitosan scaffold was tested as a proof of concept, in order to produce smart partially-biodegradable scaffolds for tissue engineering applications.
Introduction
Hydrogels are networks of hydrophilic polymers that have attracted wide research interest as controlled release devices due to their tunable chemical and three-dimensional physical structure, high water content, good mechanical properties, and biocompatibility.1Poly(N-isopropylacrylamide), PNIPAAm, is a thermoresponsive hydrogel that has low critical solution temperature (LCST), around 32 °C in an aqueous solution,2 close to body temperature. It dissolves in water below the LCST and precipitates from the aqueous solution above the LCST due to the disruption of hydrogen bonding with water and the increasing hydrophobic interactions among isopropyl groups. Due to this unique property, PNIPAAm gels have been widely used in biomedical fields, for example, as matrices in protein–ligand recognition, in drug controlled release, in enzyme and cell immobilization and in artificial organs/cell sheet technology.3
The preparation of hydrogels from soluble polymers requires typically a cross-linking procedure, that will allow the system to swell water without compromising the integrity of the material. N,N-methylenebisacrylamide (MBAM) is the cross-linking agent normally used to prepare PNIPAAm-based hydrogels. The synthesis of polyNIPAM using this cross-linker was first reported by Pelton and Chibante in 19864 and since then many publications describing the preparation, characterization and application of temperature-sensitive hydrogels have been reported in the literature.5
During the last few years, an effort has been made to modify and improve the properties of PNIPAAm hydrogels. In many attempts the strategies involve the use of organic solvents or toxic additives, that should be then recovered and recycled.6 In this paper we report the precipitation polymerization of PNIPAAm in supercritical carbon dioxide (scCO2). Our scCO2-assisted method to produce PNIPAAm hydrogels presents an enormous advantage when compared with conventional polymerization since there is no need for an intensive drying action before further processing or characterization steps.
The use of scCO2 as a polymerisation medium offers many advantages over conventional solvents: CO2 is nontoxic, non-flammable, inexpensive and readily available in high purity from a variety of sources.7 Since it is a gas at normal pressure by simply reducing the pressure of the system, it is possible to easily separate the solvent from the polymer, leading to highly pure materials8 ideal for medical applications.
The synthesis of other acrylamides in scCO2 is already reported in the literature.9 These polymer syntheses are normally performed in the presence of highly expensive surfactants to emulsify the insoluble monomer in scCO2. In our case as the monomer is highly soluble in scCO2 the polymerization could be performed without any surfactants.
Chitosan is a well known natural polymer that is biodegradable, biocompatible and non-toxic10 with possible applications in the medical field, including dialysis membranes, contact lenses, antitumor uses, drug delivery controlled-release systems and tissue engineering.
The production of smart partially-biodegradable scaffolds for tissue engineering applications combining the thermoresponsive properties of PNIPAAm, the biocompatible properties of chitosan with the green aspects of polymerizations in scCO2 is a desired goal.
In this paper we present a new strategy for the polymerization of PNIPAAm that provides new possibilities for the impregnation/coating of these hydrogels in chitosan scaffolds.
Experimental
Materials
N-isopropylacrylamide (NIPAAm, 97% purity), N,N-methylenebisacrylamide (MBAm, purity ≥ 98%), chitosan (∼85% deacetylated, medium molecular weight) and 2,2′-azobis (isobutyronitrile) (AIBN, 98% purity) were purchased from Sigma-Aldrich and used without further purification.Carbon dioxide was obtained from Air Liquide with 99.998% purity.
Preparation of PNIPAAm hydrogels
Polymerisation reactions were carried out in a high-pressure apparatus already described elsewhere.8 In a typical procedure monomer, cross-linking agent (if included), and initiator (2 wt%) are loaded into the high-pressure cell, which is then sealed and nitrogen is added to purge the cell and test leaks. The nitrogen is slowly released and liquid carbon dioxide is loaded into the cell using a high-pressure compressor. The cell is immersed in the water bath and temperature and pressure are allowed to rise to the required experimental conditions. Additional CO2 may be added to reach the exact desired pressure. The reaction was allowed to proceed for 24 hours under stirring.The reactions were performed at 65 °C and 20 MPa, according to Scheme 1. At these experimental conditions we have a homogeneous phase with all the reactants completely soluble in the supercritical medium. Different concentrations of cross-linker were tested as summarized in Table 1. Two hours after the beginning of the reaction the mixture turned yellow, then soft orange (Tyndall effect) and within a few minutes white particles of polymer started to precipitate. The resulting polymer was washed with fresh high-pressure CO2 (65 °C, 20 MPa for one hour) in order to extract the remaining residues of unreacted monomer and cross-linker. When cross-linker was added the polymers conformed closely to the interior of the reaction vessel as a dry, white polymer with a soft consistency. No significant shrinkage was observed upon venting the CO2. When no cross-linker was used the polymer precipitated at the bottom of the cell as a fluffy, dry, white, free flowing powder.
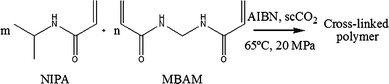 | ||
| Scheme 1 | ||
| Entry | MBAM(wt%)a | Yield(%) | LCST/°C | ΔH/J g−1 | Surface area/m2 g−1 | Polymer morphology |
|---|---|---|---|---|---|---|
| a Weight of MBAM/total weight (NIPAAm, AIBN, MBAM). | ||||||
| 1 | 0.0 | 74.7 | 33.3 | 51 | 6.1 | Highly aggregated particles |
| 2 | 1.2 | 89.6 | 31.7 | 37 | 10.6 | Defined particles slightly aggregated |
| 3 | 2.4 | 86.9 | 30.5 | 16 | 14.3 | Defined particles slightly aggregated |
| 4 | 4.5 | 94.6 | 30.7 | 10.3 | 17.3 | Defined particles slightly aggregated |
Structural/morphological analysis
The synthesized polymers were analysed by Nuclear Magnetic Resonance (NMR) and Matrix Assisted Laser Desorption/Ionization-Time of Flight Mass Spectrometry (MALDI-TOF MS). NMR spectra were performed in a Bruker equipment (ARX 400 MHz), using deuterated chloroform (CDCl3) as solvent and internal reference. MALDI-TOF MS was used to obtain the molecular weight and it was performed in an AUTOFLEX Bruker using DITHRANOL (1,8,9-anthracenetriol) as matrix. Polymer yield was determined gravimetrically.Differential scanning calorimetry (DSC) was applied to investigate the thermal features. The LCST of the hydrogel samples was determined in a PerkinElmer DSC 7. The thermal analyses were performed from 15 to 40 °C at 3 °C min−1 under a dry nitrogen atmosphere (flow rate = 20 mL min−1). Calibration for the temperature and energy scale was carried out using a pure indium standard.
Specific surface areas of the networks were determined by adsorption of N2 according to the BET method. An accelerated surface area and porosimetry system (ASAP 2010 MICROMERITICS) was used under nitrogen flow.
The morphology of hydrogel particles was also evaluated using Scanning Electron Microscopy (SEM) in a Hitachi S-2400, with an accelerating voltage set to 15 kV. Particle size diameters and porosity were obtained by image analysis using SigmaScan Pro (Systat Software Inc.). EasyFit (MathWave Technologies) software was used to fit statistical distributions to our data (Weibull, Lognormal) and perform the Kolmogorov–Smirnov and Anderson–Darling tests to select the more adequate distribution function to each system under study.11
Swelling measurements
Equilibrium hydration or swelling degree (W (%)) of synthesised samples was determined as defined by eqn (1):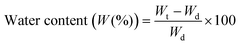 | (1) |
In situ polymerization inside chitosan scaffolds
PNIPAAm was synthesized inside chitosan scaffolds using scCO2 assisted polymerizations in order to investigate the feasibility of this technology in the production of smart partially-biodegradable matrices for tissue engineering applications.Chitosan scaffolds were previously prepared by dissolving medium molecular weight chitosan in 0.2 M acetic acid solution at a concentration of 3 wt%. The solution was then placed in cylindrical silicon moulds, freezed at −80 °C and lyophilized (Telstar-Cryodos −80, Spain) for up to 4 days to completely remove the frozen solvent. Then, the scaffolds were neutralized using a 0.1 M NaOH solution and freeze-dried again.
For the in situ polymerization of PNIPAAm, scaffolds and reactants were placed inside the high pressure vessel and scCO2 was used as carrier of the monomer/initiator/cross-linker into the porous structures as well as reaction medium. The reaction was performed under the same conditions that were tested in the preparation of PNIPPAAm hydrogels.
Results and discussion
In the absence of cross-linking agent, the precipitation polymerization of PNIPAAm in scCO2 resulted in low molecular weight polymers (MW = 660) with 75% yield (entry 1 in Table 1). Fig. 1 shows the NMR and MALDI-TOF MS spectra of PNIPAAm synthesised without cross-linker.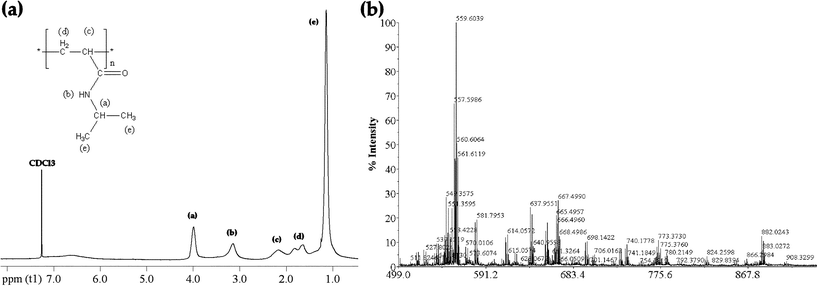 | ||
| Fig. 1 Characterization of the PNIPAAm hydrogel polymerized without cross-linker: (a) 1H NMR in CDCl3, (b) MALDI-TOF MS spectrum. | ||
The NMR confirmed the structure of PNIPAAm as well as its high purity after the efficient high-pressure CO2 washing step at the end of the reaction. The NMR and MALDI-TOF MS spectra of the synthesized cross-linked polymers were not used in the discussion due to the low solubility of these hydrogels in the solvents generally used in these techniques, such as chloroform, hexane, acetone and water. By increasing the cross-linker concentration from 1 to 5%, the yield increased up to 95% (entries 2 to 4 in Table 1). This trend might be due to the ability of the cross-linker MBAM to enhance the solubility of the growing polymer in scCO2 leading to higher yields. Cooper et al. observed a similar behaviour in the synthesis of cross-linked polystyrene in scCO2.12a
In precipitation polymerization,12 both monomer and initiator are soluble in the continuous phase but as the polymer grows the insoluble polymer chains precipitate and the particles tend to group forming an undefined, agglomerated powder. By increasing the concentration of cross-linker the number of nuclei generated in the reaction increases thus reducing the particle size diameter. Even a small increase of cross-linker (from 1 to 5%) leads to a significant decrease of the particle diameters (from 2.7 to 2.1 µm). PNIPAAm synthesized without cross-linker showed larger particles (5.4 µm), although quite agglomerated (Fig. 2(a)), while when cross-linker was added the particle diameters were significantly reduced (2.1 µm). This trend can be clearly observed by comparing SEM images from Fig. 2 and particle size distributions from Fig. 5 (see later). The small variation in cross-linker concentration explains the visual similarity between Figs. 2(b), (c) and (d). All samples show some agglomeration which is typical in a precipitation polymerization without the use of a stabiliser. Usually higher cross-linking degree results in less agglomerated polymers since particles are stabilised against coagulation by their cross-linked surfaces rather than by added stabilisers.12a This trend is also easily observed in our work. As can be seen in Fig. 2 the sample synthesised in the absence of cross-linker shows a higher degree of agglomeration when compared with the cross-linked samples. It is expected that the morphology of the materials, and their corresponding properties, could be tailored by changing the processing conditions, which makes this strategy a versatile way for preparing thermoresponsive gels.
 | ||
| Fig. 2 Scanning electron microscopy images of the hydrogels prepared under different conditions. Samples: (a) 0 wt% MBAM, (b) 1.2 wt% MBAM, (c) 2.4 wt% MBAM, (d) 4.5 wt% MBAM. | ||
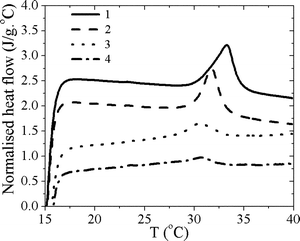 | ||
| Fig. 3 DSC thermograms of the hydrogels prepared. 1: 0 wt% MBAM; 2: 1.2 wt% MBAM; 3: 2.4 wt% MBAM; 4: 4.5 wt% MBAM. | ||
Fig. 3 shows the DSC thermograms of each sample, which were previously soaked in distilled water for 24 h. An endothermic peak is clearly observed around 32 °C, resulting from the cleavage of the hydrogen bonds between –NH and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of PNIPAAm chains and the surrounding water molecules. The values of the peak temperature (defined as LCST) and endothermic enthalpies (ΔH) are shown in Table 1. For the first three formulations a continuous decrease in the LCST is observed. We can then conclude that the cross-linking influences the phase behaviour of PNIPAAm. The strong variation in the enthalpy clearly suggests that cross-linking highly suppresses the thermoresponsive intensity of such hydrogels, as the cross-linking regions will not participate in this event, and may even repress its occurrence. It should be remarked that for linear polymers (without cross-linker) the enthalpy obtained is of the same order of previous results,2 where values of 6.3 kJ mol−1 (55.8 J g−1) were reported for PNIPPAm with different molecular weight distributions, and recognized to be typical for hydrogen bond interactions.
O groups of PNIPAAm chains and the surrounding water molecules. The values of the peak temperature (defined as LCST) and endothermic enthalpies (ΔH) are shown in Table 1. For the first three formulations a continuous decrease in the LCST is observed. We can then conclude that the cross-linking influences the phase behaviour of PNIPAAm. The strong variation in the enthalpy clearly suggests that cross-linking highly suppresses the thermoresponsive intensity of such hydrogels, as the cross-linking regions will not participate in this event, and may even repress its occurrence. It should be remarked that for linear polymers (without cross-linker) the enthalpy obtained is of the same order of previous results,2 where values of 6.3 kJ mol−1 (55.8 J g−1) were reported for PNIPPAm with different molecular weight distributions, and recognized to be typical for hydrogen bond interactions.
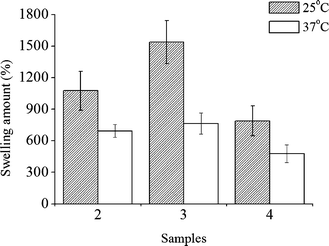 | ||
| Fig. 4 Equilibrium swelling at 37 °C and 25 °C for PNIPAAm hydrogels. 2: 1.2 wt% MBAM; 3: 2.4 wt% MBAM; 4: 4.5 wt% MBAM. | ||
The swelling degree of a thermoresponsive hydrogel is an important property to be characterised because it will determine properties such as the absorption and diffusion of solutes through the hydrogel, mechanical properties under wet conditions and water uptake capability. Fig. 4 shows the swelling behaviour, at 25 °C and 37 °C, of the PNIPAAm samples produced in scCO2.
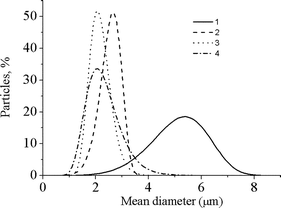 | ||
| Fig. 5 Particle size distributions of the different samples. 1: 0 wt% MBAM; 2: 1.2 wt% MBAM; 3: 2.4 wt% MBAM; 4: 4.5 wt% MBAM. | ||
In the swelling process, the water molecules penetrate the dry hydrogel, wetting the polymer chains. They are weakly adsorbed on the hydrophilic polymer chains or are connected with hydrogen bonds to each other, being trapped in the hydrophilic polymer matrices. All of these interactions lead the hydrogel to swell well at a temperature lower than the LCST. At temperatures above the LCST, this balance is disturbed, and the interactions among the hydrophobic groups begin to play a dominant role, and so the polymer chains aggregate together. As a result, the entrapped water is squeezed out. In our case 300–700% w/w differences were observed, which indicates that a considerable amount of water is entrapped inside the gel network via hydrogen bonds and influenced by the LCST. In all samples the differences in swelling for the two temperatures analysed are statistically significant with 95% confidence. The differences observed for the different samples are due to the influence of the cross-linker in the morphology of the hydrogels. As suggested by the DSC results, the water uptake should be smaller for the highest cross-linked material, being consistent with the data of Fig. 4.
The average particle size, the particle size distribution, and the porosity are also important factors in solute permeation into and out of a hydrogel. In this work the porosity of all hydrogels produced is essentially the same, around 40% even though they present quite different particle diameters and surface areas.
The surface areas obtained by the multipoint BET method (type II) are shown in Table 1. According to the isotherm analysis there is an hysteresis (H3 type) confirming an agglomerated material with slit-pores. These areas are in agreement with the particle size distribution presented in Fig. 5. The hydrogel synthesized without cross-linker has the highest mean diameter (entry 1 in Table 1), and presents the smallest surface area per gram. The particles of the hydrogels produced with different amounts of cross-linker show smaller mean diameters. There is a clear trend between the hydrogels cross-linking and surface area/particles sizes. By increasing the cross-linker concentration a decrease of particles size diameters, as well as an increase in their specific surface areas are observed. This behaviour has been observed by other authors in conventional polymerisation.13
This work demonstrates the feasibility for synthesizing thermoresponsive polymers and hydrogels in a scCO2 environment. It would be interesting to extend this strategy towards the surface modification or impregnation of previously prepared polymeric systems with a thermoresponsive polymer. This could help in preparing novel smart films, membranes or porous bulky devices with surface properties that could change with temperature. The implementation of such a strategy was tested in this work, where PNIPAAm was impregnated in situ within a biodegradable porous structure, made of chitosan, using supercritical fluid technology. Biodegradable foams can be used, for example, in tissue engineering applications where it is required as an adequate non-permanent support for cell adhesion and proliferation prior to implantation in a regenerative medicine context;14 porous biocompatible and biodegradable polymers have been widely used for this purpose. The incorporation of a fraction (mainly in the surface) of a thermoresponsive polymer could provide other possibilities to the scaffold, including improved or switchable release control of bioactive agent, such as growth factors, or to control cell adhesion/detachment. Porous structures of chitosan can be easily prepared by freeze-drying, where the final structure may be controlled by the processing conditions.15 PNIPAAm was synthesized within a previously prepared chitosan scaffold. Fig. 6 presents two SEM images of the materials prepared. Such preliminary results clearly show that the hydrogel is uniformly distributed in large amounts inside the pores of the scaffolds.
 | ||
| Fig. 6 SEM pictures of PNIPAAm hydrogel impregnated in a chitosan scaffold. | ||
Conclusion
In summary, we have developed a novel, simple and green method to synthesise PNIPAAm hydrogels without using organic solvents or any other additives (e.g. surfactants). In addition a chitosan scaffold was used as a matrix for the in situ polymerisation of NIPA in scCO2. This would allow matrices to be produced with thermoresponsive capability that could be used as porous membranes for advanced purification/separation applications, systems for switchable release of molecules or scaffolds for tissue engineering applications.Acknowledgements
We thank Professor Isabel Fonseca and Rui Viegas for help with specific surface area measurements and Professor Eurico Cabrita for helpful discussions in NMR analysis. The authors also thank the financial support from Fundação para a Ciência e Tecnologia (FCT) through contracts POCTI/35429/QUI/2000, POCTI/FIS/61621/2004, POCTI/EQU/46715/2002, SFRH/BPD/11665/2002, SFRH/BD/16908/2004 and by FEDER and FSE.References
- (a) X. Huang and T. L. Lowe, Biomacromolecules, 2005, 6, 2131 CrossRef CAS; (b) R. Langer and N. A. Peppas, AIChE J., 2003, 49, 2990 CrossRef CAS.
- (a) H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS; (b) V. V. A. Fernandez, N. Tepale, J. C. Sanchez-Diaz, E. Mendizabal, J. E. Puig and J. F. A. Soltero, Colloid Polym. Sci., 2006, 284, 387 Search PubMed.
- (a) T. Okano, N. Yamada, H. Sakai and Y. Sakurai, J. Biomed. Mater. Res., 1993, 27, 1243 CrossRef CAS; (b) P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. Chen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472 CrossRef CAS; (c) Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321 CrossRef CAS; (d) L. C. Dong and A. S. Hoffman, J. Controlled Release, 1986, 4, 223 CrossRef CAS; (e) R. A. Stile, W. R. Burghardt and K. E. Healy, Macromolecules, 1999, 32, 7370 CrossRef CAS; (f) K.-H. Park and Y. H. Bae, Biosci., Biotechnol., Biochem., 2002, 66, 1473 CrossRef CAS.
- R. Pelton and P. Chibante, Colloids Surf., 1986, 20, 247 CrossRef CAS.
- (a) S.-X. Cheng, J.-T. Zhang and R.-X. Zhuo, Cheng, S., J. Biomed. Mater. Res., 2003, 67A, 96 Search PubMed; (b) X. Zhang, Y. Yang, T. Chung and K. Ma, Langmuir, 2001, 17, 6094 CrossRef CAS; (c) R. Zhuo and W. Li, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 152 CrossRef CAS; (d) J. Zhang, S. Huang and R. Zhuo, Macromol. Chem. Phys., 2004, 205, 107 CrossRef CAS; (e) X. Zhang and R. Zhuo, Eur. Polym. Mater., 2000, 36, 2301 Search PubMed.
- (a) X.-Z. Zhang and C.-C. Chu, J. Mater. Chem., 2003, 13, 2457 RSC; (b) D. Kuckling, C. D. Vo, H.-J. P. Adler, A. Völkel and H. Cölfen, Macromolecules, 2006, 39, 1585 CrossRef CAS.
- (a) A. I. Cooper, J. Mater. Chem., 2000, 10, 207 RSC; (b) J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543 CrossRef CAS.
- (a) T. Casimiro, A. M. Banet-Osuna, A. M. Ramos, M. Nunes da Ponte and A. Aguiar-Ricardo, Eur. Polym. J., 2005, 41, 1947 CrossRef CAS.
- F. A. Adamsky and E. J. Beckman, Macromolecules, 1994, 27, 312 CrossRef CAS; (a) W. Ye and J. M. DeSimone, Macromolecules, 2005, 38, 2180 CrossRef CAS.
- (a) P. J. VandeVord, H. W. T. Matthew, S. P. DeSilva, L. Mayton, B. Wu and P. H. Wooley, J. Biomed. Mater. Res., 2002, 59, 585 CrossRef CAS; (b) K. Tuzlakoglu, C. M. Alves, J. F. Mano and R. L. Reis, Macromol. Biosci., 2004, 4, 811 CrossRef CAS; (c) A. Di Martino, M. Sittinger and M. V. Risbud, Biomaterials, 2005, 26, 5983 CrossRef.
- M. Temtem, T. Casimiro and A. Aguiar-Ricardo, J. Membr. Sci., 2006, 283, 244 CrossRef CAS.
- (a) A. I. Cooper, W. P. Hems and A. B. Holmes, Macromolecules, 1999, 32, 2156 CrossRef CAS; (b) R. Arshady, Colloid Polym. Sci., 1992, 270, 717 CrossRef CAS; (c) W. Wang, R. M. T. Griffiths, A. Naylor, M. R. Giles, D. J. Irvine and S. M. Howdle, Polymer, 2002, 43, 6653 CrossRef CAS.
- C. Sayil and O. Okay, Polym. Bull., 2002, 48, 499 Search PubMed.
- (a) M. Chapekar, J. Biomed. Mater. Res. (Appl. Biomater.), 2000, 53, 617 Search PubMed; (b) X. Liu and P. X. Ma, Ann. Biomed. Eng., 2004, 32, 477 CrossRef; (c) V. Karageorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474 CrossRef CAS.
- S. V. Madihally and H. W. T. Matthew, Biomaterials, 1999, 20, 1133 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |