Separation of aromatic hydrocarbons from alkanes using the ionic liquid 1-ethyl-3-methylimidazoliumbis{(trifluoromethyl) sulfonyl}amide†
Alberto
Arce
a,
Martyn J.
Earle
*b,
Héctor
Rodríguez
a and
Kenneth R.
Seddon
b
aDepartment of Chemical Engineering, University of Santiago de Compostela, E-15782, Santiago de Compostela, Spain
bThe QUILL Research Centre, The Queen's University of Belfast, Belfast, BT9 5AG, UK. E-mail: quill@qub.ac.uk
First published on 25th October 2006
Abstract
The liquid–liquid equilibrium for the ternary system formed by hexane, benzene and the ionic liquid 1-ethyl-3-methylimidazoliumbis{(trifluoromethyl)sulfonyl}amide, [C2mim][NTf2], has been experimentally determined at 25 °C and 40 °C. The results show that the [C2mim][NTf2] can selectively remove benzene from its mixtures with hexane, suggesting that this ionic liquid can be used as an alternative solvent in liquid extraction processes for the removal of aromatic compounds from their mixtures with alkanes.
Introduction
Separation of aromatic and non-aromatic compounds has traditionally been one of the most challenging tasks in refinery processes.1 Their boiling points are often rather close, making it difficult to perform the separation by simple distillation under economically acceptable conditions. In addition, the formation of azeotropes may occasionally complicate the operation even more.2The common processes usually chosen by industry to deal with such separations are liquid extraction, extractive distillation and azeotropic distillation.2,3 Each of these techniques is suitable for the treatment of feeds within different ranges of aromatic contents. Typically this ranges from 20 to 100 wt%, with none of them showing favourable economics to manage feeds below 20 wt% of aromatics.3 Liquid extraction is currently the most interesting alternative to process hydrocarbon streams, with an aromatic content in the range 20 to 65 wt%. Compared to both extractive and azeotropic distillation techniques, liquid extraction has a low energetic demand, so it would be valuable to extend the range in which extraction might be profitably used.
An inherent problem to all the three techniques cited above is related to use of a solvent, which is mixed with the feed and comes out of the extractor, or the column, accompanying the raffinate and extract streams. Typical solvents in these processes are organic compounds such as sulfolane (conventionally the principal one), ethylene glycols, dimethylsulfoxide, N-methylpyrrolidone, N-formylmorpholine or carbonate derivates, among some others.2–4 The recovery of these solvents from the extract and raffinate streams must be carried out by distillation, thus resulting in an additional increase in the installation and operational costs of the processes.
Apart from the thermodynamically favourable behaviour for the extraction, desirable attributes for the solvents are: chemical stability, low toxicity, non-corrosivity, low cost, and ease in their recovery from the extract.5 Considering this list, ionic liquids might constitute an alternative option for conventional solvents in liquid extraction.6,7 Room temperature ionic liquids have received increasing attention by the scientific community, particularly since the late 1990s.8 They have a negligible vapour pressure at ambient temperatures and pressures,9 which would open the possibility of using simpler techniques, such as flash distillation, in the recovery of the ionic liquids from the raffinate and extract streams.‡ In addition, and despite several exceptions, there is a wide variety of chemically stable, non-corrosive ionic liquids, with most of them possessing a large liquid range. Their current problems could be the high price and the relatively unknown toxicity,10 the former being expected to dramatically decrease as a result of their implementation in industrial processes, and their production at large scale, and the later requiring further research to establish standard assessments.11
Several groups have already explored the application of ionic liquids to liquid extraction,12 with the separation of aromatic and non-aromatic hydrocarbons being a key focus. However, only a few papers report the liquid–liquid equilibrium (LLE) data of the systems studied.7,13–15
Here we present the LLE data of the ternary system (hexane + benzene + [C2mim][NTf2]) at two different temperatures, 25 °C and 40 °C, in the classical range of temperatures in which extraction at atmospheric pressure has a practical application. Hexane and benzene have been chosen as representatives of aliphatic and aromatic compounds. The ionic liquid has been preliminary selected according to its low melting point (well below room temperatures), its relatively low viscosity (thus facilitating fluid flow and mass transfer), and the fact that the anion [NTf2]– does not decompose to give HF under conditions normally used for liquid–liquid extractions, as some other typical anions in ionic liquids chemistry do.16
Experimental
Benzene with a nominal purity of 99.0% was purchased from LabScan. Hexane with a nominal purity of 97.0% was supplied by BDH. Both hydrocarbons were used as received, without further purification. The ionic liquid [C2mim][NTf2] was prepared by a metathetic reaction of [C2mim]Cl and Li[NTf2], following an analogous procedure to that described elsewhere;17 the water content of the final product was found to be <100 ppm, quantified by Karl-Fischer titration.18The feasibility of [C2mim][NTf2] as solvent for the liquid–liquid extraction of benzene from its mixture with hexane was analyzed by determining experimental tie-lines covering the whole immiscibility region of the ternary system (hexane + benzene + [C2mim][NTf2]). Mixtures of all the three compounds (or just two, in the cases of the two immiscible pairs of the system) with a global composition being in the immiscibility region, were prepared. These mixtures were placed inside a glass cell, sealed, and thermostatically controlled at 25.0 °C or 40.0 °C. The mixtures were vigorously stirred for not less than 1 h, followed by a settling period of several hours to ensure that the thermodynamic equilibrium was reached. Next a sample was taken from each equilibrium phase into which the system had split: a top (hydrocarbon-rich) phase and a bottom (ionic liquid-rich) phase, and the compositional analyses were performed as indicated below.
The determination of the composition of the samples was carried out by means of 1H NMR spectroscopy. Details of the specific method, already applied in the study of LLE ternary systems with ionic liquids, can be found elsewhere.19 This technique combines simplicity and sufficient accuracy for practical purposes. To assess the precision of the method in this particular case, homogeneous probe vials with overall compositions in the proximities of the binodal curve, and covering the whole range of compositions, were prepared by weight using a Metler Toledo AT261 balance, precise to within ±10–4 g. After dissolving the samples in deuterated solvent and running them in a Bruker Avance DPX-500 spectrometer, selected peaks for each component were integrated to calculate the molar fractions. The measured results were found to be in good agreement with the actual compositions. Calibration lines with standard deviations of 0.006 or less for each compound were obtained. The maximum absolute deviation was found to be 0.009 in mole fractions. Thus, the technique was validated and used within the equilibrium experiments.
Results and discussion
The measured compositions of the experimental tie-line ends of the ternary system (hexane + benzene + [C2mim][NTf2]) at 25 °C are reported in Table 1; the data corresponding to a temperature of 40 °C are shown in Table 2.| Hydrocarbon-rich phase | Ionic liquid-rich phase | β | S | ||||
|---|---|---|---|---|---|---|---|
| x 1 | x 2 | x 3 | x 1 | x 2 | x 3 | ||
| 1.000 | 0.000 | 0.000 | 0.039 | 0.000 | 0.961 | — | — |
| 0.930 | 0.070 | 0.000 | 0.038 | 0.096 | 0.866 | 1.37 | 33.56 |
| 0.834 | 0.166 | 0.000 | 0.038 | 0.211 | 0.751 | 1.27 | 27.90 |
| 0.717 | 0.283 | 0.000 | 0.035 | 0.327 | 0.638 | 1.16 | 23.67 |
| 0.573 | 0.427 | 0.000 | 0.039 | 0.432 | 0.529 | 1.01 | 14.86 |
| 0.431 | 0.569 | 0.000 | 0.034 | 0.523 | 0.443 | 0.92 | 11.65 |
| 0.300 | 0.700 | 0.000 | 0.031 | 0.597 | 0.372 | 0.85 | 8.25 |
| 0.112 | 0.888 | 0.000 | 0.018 | 0.692 | 0.290 | 0.78 | 4.85 |
| 0.000 | 1.000 | 0.000 | 0.000 | 0.757 | 0.243 | 0.76 | — |
| Hydrocarbon-rich phase | Ionic liquid-rich phase | β | S | ||||
|---|---|---|---|---|---|---|---|
| x 1 | x 2 | x 3 | x 1 | x 2 | x 3 | ||
| 1.000 | 0.000 | 0.000 | 0.041 | 0.000 | 0.959 | — | — |
| 0.877 | 0.123 | 0.000 | 0.038 | 0.148 | 0.814 | 1.20 | 27.77 |
| 0.733 | 0.267 | 0.000 | 0.039 | 0.291 | 0.670 | 1.09 | 20.48 |
| 0.590 | 0.410 | 0.000 | 0.038 | 0.419 | 0.543 | 1.02 | 15.87 |
| 0.449 | 0.551 | 0.000 | 0.036 | 0.503 | 0.461 | 0.91 | 11.39 |
| 0.303 | 0.697 | 0.000 | 0.032 | 0.578 | 0.390 | 0.83 | 7.85 |
| 0.233 | 0.767 | 0.000 | 0.030 | 0.618 | 0.352 | 0.81 | 6.26 |
| 0.106 | 0.894 | 0.000 | 0.018 | 0.699 | 0.283 | 0.78 | 4.60 |
| 0.000 | 1.000 | 0.000 | 0.000 | 0.764 | 0.236 | 0.76 | — |
The feasibility of using the ionic liquid as a solvent to perform the extraction of benzene from a mixture with hexane was evaluated by classic parameters such as the solute distribution ratio (β) and the selectivity (S), calculated from the experimental data. These parameters are defined by the following expressions:
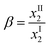 | (1) |
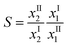 | (2) |
Fig. 1(a) and (b) show graphical representations of the tie-lines of the ternary systems at 25 °C and 40 °C, respectively, in a triangular diagram. As it can be seen (Tables 1 and 2), the effect of the temperature is rather small, with the characterising parameters of the extraction actually being slightly better for the lower temperature. Therefore, the quality of the process is maximised by working at room temperature, thus avoiding the consumption of energy required to heat the extracting unit.
![(a) Experimental tie-lines (○, solid line) for the LLE of the ternary system (hexane + benzene + [C2mim][NTf2]) at 25 °C. The corresponding tie-lines correlated by means of the NRTL equation are also plotted (□, dotted line). (b) Experimental tie-lines (○, solid line) for the LLE of the ternary system (hexane + benzene + [C2mim][NTf2]) at 40 °C. The corresponding tie-lines correlated by means of the NRTL equation are also plotted (□, dotted line).](/image/article/2007/GC/b610207g/b610207g-f1.gif) | ||
| Fig. 1 (a) Experimental tie-lines (○, solid line) for the LLE of the ternary system (hexane + benzene + [C2mim][NTf2]) at 25 °C. The corresponding tie-lines correlated by means of the NRTL equation are also plotted (□, dotted line). (b) Experimental tie-lines (○, solid line) for the LLE of the ternary system (hexane + benzene + [C2mim][NTf2]) at 40 °C. The corresponding tie-lines correlated by means of the NRTL equation are also plotted (□, dotted line). | ||
By inspection of the LLE data (or the triangular diagrams), another significant aspect is the fact that the ionic liquid does not enter the equilibrium hydrocarbon-rich phase. At least, the presence of [C2mim][NTf2] was not detected in the upper phase by NMR. In an attempt to measure presence of traces of [C2mim][NTf2] in the raffinate stream, two mixtures of [C2mim][NTf2] with either hexane (250 cm3) or a 15% benzene/85% hexane mixture (250 cm3) were prepared and after equilibrating at 25 °C, the [C2mim][NTf2] layer was separated. The solvent was evaporated and the residues were weighed. The hexane layer contained no measurable ionic liquid (<0.1 ppm) and the 15% benzene/85% hexane mixture was found to contain 1.4 ppm ionic liquid. This showed that a post-extraction treatment of the raffinate to recover the solvent and turn it back to the extraction unit, may be necessary if the benzene content of the raffinate was high.
The graphical representation of the solute distribution ratio, at 25 °C, as a function of the solute mole fraction in the hydrocarbon-rich phase is shown in Fig. 2. A comparison with data in literature20 for the ternary system (hexane + benzene + sulfolane {2,3,4,5-tetrahydrothiophene-1,1-dioxide}) at the same temperature is made. At present sulfolane is the most widely recognised solvent in industrial processes for the separation of aromatic and aliphatic hydrocarbons by liquid extraction. The values of β in the comparable zone are clearly higher when using the ionic liquid, thus indicating a clear preference of the ionic liquid for the benzene in the mixture of hydrocarbons, and also meaning a reduction in the amount of solvent required to carry out the treatment of a given feed stream.
![Solute distribution ratio for the ternary systems (hexane + benzene + [C2mim][NTf2]) (●) and (hexane + benzene + sulfolane) (○), at 25 °C, as a function of the mole fraction of benzene in the hydrocarbon-rich phase.](/image/article/2007/GC/b610207g/b610207g-f2.gif) | ||
| Fig. 2 Solute distribution ratio for the ternary systems (hexane + benzene + [C2mim][NTf2]) (●) and (hexane + benzene + sulfolane) (○), at 25 °C, as a function of the mole fraction of benzene in the hydrocarbon-rich phase. | ||
As the solute distribution ratios with the ionic liquid decrease from values above the unity to values below it (equivalently, there is a change in the sign of the tie-lines slope), the system presents solutropy.21 The solutrope corresponds to the horizontal tie-line for which β = 1; in this case, it happens for immiscible mixtures with an overall mole fraction of benzene somewhere in the range 0.40–0.50. To date, there are only a few values of the solute distribution ratio above unity reported in literature, for the separation of aromatic and aliphatic hydrocarbons.
Selectivity for the data at 25 °C was also plotted as a function of the mole fraction of solute in the organic phase, and compared to the system with sulfolane as solvent.20 The results are shown in Fig. 3. The selectivity for the sulfolane system is higher at the lowest values of aromatic hydrocarbon content, then decreasing faster than the selectivity with the ionic liquid as the mole fraction of benzene in the system increases. Thus, the sulfolane's only advantage over the [C2mim][NTf2] is in a narrow range. Nevertheless, as it was stated in the introductory section of this paper, at such low levels of aromatic hydrocarbon content, the extraction process is not viable from an economical point of view when using volatile compounds such as sulfolane, due to the cost of the post-extraction stages for the recovery of the solvent. The problem might be avoided by using the ionic liquid, as its negligible vapour pressure makes it easier to recover. Therefore, from this perspective, [C2mim][NTf2] is also more advantageous than sulfolane in practical processes.
![Selectivity for the ternary systems (hexane + benzene + [C2mim][NTf2]) (●) and (hexane + benzene + sulfolane) (○), at 25 °C, as a function of the mole fraction of benzene in the hydrocarbon-rich phase.](/image/article/2007/GC/b610207g/b610207g-f3.gif) | ||
| Fig. 3 Selectivity for the ternary systems (hexane + benzene + [C2mim][NTf2]) (●) and (hexane + benzene + sulfolane) (○), at 25 °C, as a function of the mole fraction of benzene in the hydrocarbon-rich phase. | ||
Data correlation
The correlation of the LLE data is interesting as it permits a computational treatment and opens the possibility of designing simulation software. Thus, in this case, the non-random two-liquid (NRTL) equation22 was used to correlate the experimental data. In spite of being a model initially developed for non-electrolytes, previous works13–15,19 demonstrate that the original NRTL equation can successfully correlate LLE data of ternary systems involving ionic liquids.A computer program by Sørensen23 was used to calculate the binary interaction parameters of the NRTL equation. A summary of how this program calculates the binary interaction parameters of the model can be found elsewhere.19 The capability of the correlation was evaluated by means of the residual function F and the mean error of the solute distribution ratio Δβ, defined as:
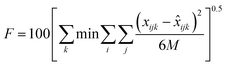 | (3) |
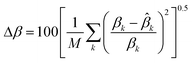 | (4) |
The non-randomness parameter of the model, α, was set to different values between 0.10 and 0.40, the best results being found for the correlation with α equal to 0.32, both at 25 °C and at 40 °C.22 The binary interaction parameters, as well as the values of the corresponding residuals, are reported in Table 3. As expected, the difference between the parameters sets for each temperature is small. The correlated tie-lines are plotted in Fig. 1(a) and Fig. 1(b), along with the experimental ones. In general, the correlation is quite accurate. However, the major problem of the model is that it can not properly correlate the absence of ionic liquid in the hydrocarbon-rich phase.
| T/°C | Compounds | Binary interaction parameters (α = 0.32) | F | Δβ | |
|---|---|---|---|---|---|
| i–j | Δgij/kJ mol–1 | Δgji/kJ mol–1 | |||
| 25.0 | 1–2 | –2.5216 | 5.0452 | 0.1790 | 2.6 |
| 1–3 | 12.020 | 6.7120 | |||
| 2–3 | 15.102 | –3.2487 | |||
| 40.0 | 1–2 | –2.7282 | 5.0288 | 0.3570 | 3.0 |
| 1–3 | 12.666 | 7.0304 | |||
| 2–3 | 15.092 | –3.4147 | |||
Conclusions
The experimental determination of the LLE of the ternary system (hexane + benzene + [C2mim][NTf2]) at 25 °C and 40 °C has been carried out. By analysing the data, it has been demonstrated that [C2mim][NTf2] can be an excellent solvent for liquid extraction processes for the separation of aromatic and aliphatic hydrocarbons. The values of the solute distribution ratio and the selectivity are good, being better than those for sulfolane, which is the conventional solvent of reference for these purposes. The [C2mim][NTf2] is an ionic liquid with a melting point below room temperature, with a relatively low viscosity, and does not present problems of decomposition or corrosion.8,11As the ionic liquid is present in minute quantities in the equilibrium upper phase, the treatment of the raffinate stream to recover the solvent accompanying the hydrocarbons may be unnecessary. In addition, due to the negligible vapour pressure of the ionic liquids, these solvent recovery operations are easier than when using conventional organic solvents. Therefore, the use of the ionic liquids can lead to major savings in the installation and operational costs of the processes. Also, the economical feasibility of the liquid extraction to perform the separation of aromatics and aliphatics may take place under a wider range of feed conditions than are currently used.
A very small effect of temperature has been observed, the results being slightly better for the lower temperature. Hence, the efficiency of the extraction by [C2mim][NTf2] is better at room temperature, not requiring energy to keep a higher temperature in the extractor.
The main handicap of [C2mim][NTf2] towards implementation in real processes might be its current high price; however, this price is expected to critically decrease in the near future as it will be produced on a much larger scale. Moreover, there is plenty of scope to explore other hydrophobic or hydrophilic ionic liquids for this process; there is nothing optimal in the choice of [C2mim][NTf2]—it is simply an exemplar.
Note added in proof
Since submitting this manuscript, two relevant papers have appeared (ref. 24 and 25). These data complement the results presented here.Acknowledgements
HR wants to thank The QUILL Centre for hosting him as a temporary research fellow, and express his gratitude to the Ministerio de Educación y Ciencia (Spain) for the FPI grant with reference BES-2004-5311 under project PPQ2003-01326. We would also like to acknowledge the QUILL Industrial Advisory Board for their support.References
- U. Razdan, S. V. Joshi and V. J. Shah, Curr. Sci., 2003, 85, 761–771 CAS.
- A. A. Gaile, G. D. Zalishchevskii, N. N. Gafur, L. V. Semenov, O. M. Varshavskii, N. P. Fedyanin and L. L. Koldobskaya, Chem. Technol. Fuels Oils, 2004, 40, 215–221 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, VCH, New York, USA, 2nd edn, 1993, pp. 316–320 Search PubMed.
- R. Wennersten, in Principles and practices of solvent extraction, ed. J. Rydberg, C. Musikas and G. R. Choppin, Marcel Dekker, New York, USA, 1992, p. 328 Search PubMed.
- D. M. T. Newsham, in Science and Practice of Liquid–Liquid Extraction, ed. J. D. Thornton, Clarendon Press, Oxford, UK, 1992, vol. 1, pp. 27–31 Search PubMed.
- G. W. Meindersma, A. J. G. Podt and A. B. de Haan, Fuel Process. Technol., 2005, 87, 59–70 CrossRef.
- N. Deenadayalu, K. C. Ngcongo, T. M. Letcher and D. Ramjugernath, J. Chem. Eng. Data, 2006, 51, 988–991 CrossRef CAS.
- Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2003 Search PubMed.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS.
- J. Ranke, K. Mölter, F. Stock, U. Bottin-Weber, J. Poczobutt, J. Hoffmann, B. Ondruschka, J. Filser and B. Jastorff, Ecotoxicol. Environ. Saf., 2004, 28, 396–404 CrossRef.
- R. P. Swatloski, J. D. Holbrey, S. B. Memon, G. A. Caldwell, K. A. Caldwell and R. D. Rogers, Chem. Commun., 2004, 668–669 RSC.
- U. Domańska, A. Pobudkowska and F. Eckert, Green Chem., 2006, 8, 268–276 RSC.
- M. S. Selvan, M. D. McKinley, R. H. Dubois and J. L. Atwood, J. Chem. Eng. Data, 2000, 45, 841–845 CrossRef CAS.
- T. M. Letcher and N. Deenadayalu, J. Chem. Thermodyn., 2003, 35, 67–76 CrossRef CAS.
- T. M. Letcher and P. Reddy, J. Chem. Thermodyn., 2005, 37, 415–421 CrossRef CAS.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- P. Bonhôte, A.-P. Diaz, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef CAS; P. Bonhôte, A.-P. Diaz, M. Armand, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1998, 37, 166 CrossRef CAS.
- A. Stark, M. J. Torres and K. R. Seddon, Pure Appl. Chem., 2000, 72, 2275–2287 CrossRef CAS.
- A. Arce, O. Rodríguez and A. Soto, J. Chem. Eng. Data, 2004, 49, 514–517 CrossRef CAS.
- J. Chen, L.-P. Duan, J.-G. Mi, W.-Y. Fei and Z.-C. Li, Fluid Phase Equilib., 2000, 173, 109–119 CrossRef CAS.
- J. P. Novák, J. Matous and J. Pick, Liquid–Liquid Equilibria, Elsevier, Amsterdam, 1987, pp. 128–129 Search PubMed.
- H. Renon and J. M. Prausnitz, AIChE J., 1968, 14, 135–144 CrossRef CAS.
- J. M. Sørensen, doctoral thesis, Danmarks Tekniske Højskole, Lingby, Denmark, 1980.
- G. W. Meindersma, A. J. G. Podt and A. B. de Haan, Fluid Phase Equilib., 2006, 247, 158–168 CrossRef CAS.
- G. W. Meindersma, A. Podt and A. B. de Haan, J. Chem. Eng. Data, 2006, 51, 1814–1819 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Calculation of compositions of phases at equilibrium. See DOI: 10.1039/b610207g |
| ‡ Although it is known that ionic liquids can be distilled at high temperature and low pressure,9 under the conditions envisaged here they may be considered as essentially non-volatile. |
| This journal is © The Royal Society of Chemistry 2007 |