Detection of sulfur-free odorants in natural gas using ion mobility spectrometry
V.
Ruzsanyi
a,
S.
Sielemann
a and
J. I.
Baumbach
b
aG.A.S. Gesellschaft für analytische Sensorsysteme mbH, Otto-Hahn-Str. 13, 44227, Dortmund, Germany
bISAS – Institute for Analytical Sciences, Bunsen-Kirchhoff-Straße 11, 44139, Dortmund, Germany
First published on 12th December 2006
Abstract
Beside the primary motivation of the public gas suppliers for odorizing natural gas with a sulfur-free odorant, which relates to the image of the environment-friendly fuel, natural gas, competing with low-sulfur heating fuel and diesel, a question of crucial importance of how to detect such sulfur-free odorants comes up. Concerning the replacement of sulfur-containing by sulfur-free odorization, the availability of a fast and sensitive detection method that can, further, be used on-site plays a key role. The minimum concentration of the new sulfur-free odorant Gasodor™ S-Free™ (S-Free) in natural gas should be added at a level of at least 8.8 mg m−3 to assure a significant warning smell. Therefore, a dynamic range between 0 and approx. 25 mg m−3 must be realised in the rather complex matrix of natural gas. By means of a handheld ion mobility spectrometer, the odorant content in natural gas is determined within less than 80 s total analysis time directly at the gas pipe. The concentration of S-Free is monitored between 4 and 23 mg m−3 respecting the quality of the natural gas (high- and low-caloric gas). Results of the validation using a gas chromatograph as a reference standard will be discussed in detail.
Introduction
Due to image and competition reasons, the gas industry intends to introduce a sulfur-free odorant for natural gas to keep sulfur dioxide emissions to the atmosphere as low as possible. In Germany, sulfur containing odorization leads to additional sulfur dioxide emissions in the range of approx. 400–600 t per year.1Nevertheless, natural gas has the lowest sulfur content compared to other primary energy sources. Using sulfur-free odorants could reduce the emissions further, because the odorization is an additive to natural gas, required at a certain security level that assures a significant warning smell.
Actually, based on the results of preliminary investigations on smell character, analytics and stability, and after successfully passing type test according to DIN 30561, a mixture of methylacrylate (37.4 mass%), ethylacrylate (60.0 mass%) and methylethylpyrazine (2.6 mass%) as stabilizer has a good chance to substitute sulfur containing odorants in the near future. This odorant is registered under the trade name Gasodor™ S-Free™.2
The requirements of DIN EN ISO 13734 were totally fulfilled with the natural restriction that the odorant is not based on sulfurous compounds. The sulfur-free odorant is suitable for application in quantity-dependent controlled dosing equipment and appropriate to the requirements of the DVGW, the German Technical and Scientific Association for Gas and Water. Some field tests have shown that the availability of fast and reliable detection systems to assure a sufficient odour content in natural gas plays a key role in the acceptance of the odorant for the gas supplying companies. Field tests confirmed that, in comparison to sulfur-containing odorants, the acrylates are less adsorbed in the distribution grid. Furthermore, the sulfur-free odorant is highly economical, even when central odorization in larger networks is applied. Moreover, the corrosion potential of the flue gas in the combustion process can be reduced.2–10
The supervision on odorization stations, as well as at the different gas net points, must be realized in a reliable and efficient way. Generally, for the detection of the odorant level in natural gas, chromatographic methods (e.g. micro GC (μGC) by Varian or Agilent) and olfactometric instruments (e.g. Olfactomer DTEX by Axel Semrau) are used.11,12 Recently, a method based on the interpretation of colour changes by chemical reactions within a reaction tube was also developed (tubes and CMS-chips).13 Concerning these detection techniques, major differences are to be considered comparing detection limit, total analysis time and price level. Generally the total analysis time is less than 10 minutes. The difference in the accuracy of the various detection methods is rather high. GCs achieve an accuracy of approx. 10%, whereas in the case of tubes measurement errors in the range of up to 30% are specified by the producers.
In order to control the concentration level, handheld equipment is generally preferred from the utility’s side, with the exception of stationary instruments applied for continuous odorization control at fixed points. The control of measuring points situated far from the odorization point and on the border of the local gas net is particularly important because the odorant concentration at these points greatly depends on the gas consumption. In this case the use of mobile, handheld devices is required. Furthermore, these have to be adapted to harsh outdoor conditions and not need special handling and transportation or operators trained in analytics.
Today’s process gas chromatographs with thermal conductivity detector (TCD) and ion mobility spectrometers (IMS) could fulfil the needs of reliable surveillance. In some cases laboratory GC-systems can contain so called differential mobility detectors (DMD), a special kind of ion mobility spectrometer.1,11,12 It will be shown that such a pre-separation of the analytes before entering the detector using the gas chromatographic columns is not indispensable. A handheld ion mobility spectrometer with membrane inlet system will be described, and it will be shown that the automatic interpretation of the relevant parts of the spectra, obtained with respect to the monitoring of the odorant level by the sulfur-free odorants, on the basis of spectra series could be realised effectively. In the present case, the instrumentation based on IMS is about 10 times smaller than the portable GC-systems. As for its operation, no heating, valve system or gas tank is required.
Instrumentation and method
The applied technology, ion mobility spectrometry, is based on the characterization of analytes through their gas phase ion mobility. Basic working principles of the method are described in the literature in detail and will not be mentioned here.14–19In the present application concerning the criteria of a rapid, in situ quantification of S-Free in different gas stations, a handheld instrument was developed based on a miniaturised IMS (μIMS). The μIMS is a commercially available instrument (G. A. S. Gesellschaft fuer analytische Sensorsysteme mbH, Dortmund) specially adapted for the detection of S-Free in the concentration range of 4 to 23 mg m−3. Details of the instrumentation set-up and the developed method will be presented in this and the next section.
The portability of the instrument is assured through an internal gas circuit, which consists of a membrane inlet system coupled with a valve-switching, and, as well as a pump for introducing the sample gas, a gas filter to keep the moisture content low enough and for filtering other contaminations coming from the sample gas, and a second pump for transporting the gas into the circuit (Fig. 1).
 | ||
| Fig. 1 Set-up of the μIMS. | ||
The sampling of the natural gas is realized directly through a by-pass gas stream using pump 1. To avoid the influence of a possible change of pressure within the gas net, the flow rate of the natural gas stream in the by-pass is regulated and kept constant using a digital flow meter, which can be applied between 0.02–10 bar. During the sampling, the gas molecules permeate through the membrane and reach the internal gas circuit where they are carried through the sample gas stream directly into the ionisation room of the IMS before being analysed. To gain a stable permeation rate, the temperature of the membrane is held constantly at 40 °C. At the end of the sampling process, a switch of the valve assures the absorption of ambient air to clean the membrane.
One measurement cycle, including pre-cleaning of the membrane, sampling, analysis and further cleaning of the system, takes approx. 80 seconds.
The calibration of the instruments was investigated in different types of natural gases. Certified test gases with an S-Free concentration higher than 25 mg m−3 were diluted with the same type of natural gas without odorant using mass flow controllers (red-y, Vögtlin Instruments AG, Switzerland) to examine the whole range of interest (4 to 23 mg m−3). The different odorant concentrations were realized in 10 and 20 per cent dilution steps. Each concentration level was analysed three times to prove the reproducibility of the devices. The adjusted concentration levels were controlled using a μGC (3000 A, Agilent Technologies) in combination with a thermodesorption unit (Micro TD, Airsense Analysetechnik GmbH).
Results
The intention of the present application is the detection and quantification of the major components of the sulfur-free odorant, methylacrylate and ethylacrylate, directly within the natural gas as a short-time method without gas chromatographic pre-separation. The method is generally based on the effective and selective ionisation of the acrylates in the natural gas matrix.The kind of ions produced in IMS differs depending on the method of ionisation. By using a radioactive ionisation source the carrier gas molecules are ionised by the β-particles directly.
Positive carrier gas ions and free electrons will become available. Through further reaction with the moisture and other impurities, such as NH3 and NO existing at low level in the pure drift gas and sample gas, so called reaction ions will be formed. These reaction ions will undergo chemical reactions with the analyte molecules to build so called analyte ions. For this process, the proton affinity of the analyte has to be higher than the proton affinity of water (692 kJ mol−1) using positive drift voltage polarity. Consequently, for the successful detection of an analyte in a complex mixture the proton affinity plays the key role due to the different competitive ion–molecule reactions in the ionisation room of the IMS.
Thus, in the present case, the effective ionisation of the odorant in the natural gas matrix is secured due to the high difference between the proton affinities of the acrylates and other compounds (mainly methane) of the natural gas.
To identify the characteristic peaks in the spectrum of S-Free in natural gas, a spectrum of a mixture of methylacrylate and ethylacrylate in the same mixing ratio as it is in S-Free in ambient air was used. The spectrum of the mixture is shown in Fig. 2. With a reaction ion peak (RIP) at 2.86 ms, the spectrum shows five additional signals at drift times of 3.07, 3.23, 3.65, 3.85 and 4.04 ms, with calculated reduced mobilities of 3.57, 3.39, 3.00, 2.85 and 2.71 cm2 V−1 s−1, respectively. The peaks presumably correspond to the monomer ion of methylacrylate (1) and ethylacrylate (2), the dimer ion of methylacrylate (3) and ethylacrylate (5) and a mixed dimer ion of methylacrylate and ethylacrylate (4).
 | ||
| Fig. 2 Spectrum of a mixture of methylacrylate and ethylacrylate. | ||
The characteristic IMS spectra of S-Free in natural gas is shown in Fig. 3 at two concentration levels. The comparison of the spectrum of the mixture of the pure compound (Fig. 2) with the spectrum of S-Free (Fig. 3) shows that there are no additional signals due to the components of natural gas itself. Also, the stabilizer methylethylpyrazine is not visible.
 | ||
| Fig. 3 Characteristic spectrum of S-Free. | ||
The reaction ion peak (RIP) is at 2.87 ms; the major characteristic peak of S-Free is at 4.13 ms, and is therefore due to the main compound ethylacrylate.
Comparing the spectra obtained at low (5.4 mg m−3) and at high (21.6 mg m−3) odorant level it can be observed that the RIP decreases, whereas the peak at 4.13 ms increases with higher S-Free concentration because of the ion–molecule reactions (proton transfer) between the reaction ions and analytes. Thus, the tracking of changes of these two peaks selected, RIP and AIP (analyte ion peak) will lead to sufficient and significant determination of the concentration of this odorant in natural gas.
Fig. 4 allows a better visualisation of the changes of the ion mobility spectra during the whole analysis sequence. At the beginning of the measurement, before sampling and in the absence of the acrylates, just the reaction ions are formed in air so that only the peak of the reaction ions can be seen. In the case of sampling of 20 spectra, when natural gas permeates through the membrane inlet system into the IMS, the peak of S-Free is formed. After reaching the maximum peak height (following the axis “Number of Spectrum”) the signal is reduced, due to the flushing out of the analytes.
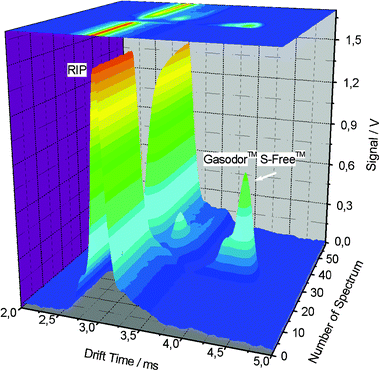 | ||
| Fig. 4 3D-IMS plot of the analysis of S-Free. | ||
In the case of the maximum peak height, almost all charges are transferred from the reactions ions to the analytes, visible as an apparent absence of the RIP near spectrum number 30 while analysing a sample with higher odorant concentration, such as 21.6 mg m−3. This occurs according to the optimisation of the experimental parameters of the analysis, so that the complete concentration range of interest (up to 23 mg m−3) can be covered. It becomes obvious that a characterization of the odorants can successfully be achieved in both ways, either using the position of the peak on the drift time scale or regarding the peak intensity as function of drift time and running time of the experiment related to the introduction and flushing out of analytes in the peak-height diagram.
It is a fact that natural gases differ from one another with respect to their quality and composition, which mainly depends on their provenance.
The distinction of the gas types is mainly characterized by the caloric value and designated as so-called L- or H-gas (“L” stands for low and “H” for high caloric). The composition correlates with the caloric value; H-gases generally contain a higher amount of methane and lower amount of nitrogen. Correspondingly L-gases typically carry various hydrocarbons with longer chains (C4–C6). Therefore L-gases can be considered as a more inhomogeneous matrix regarding the various proton affinities of the compounds.
Fig. 5 shows a characteristic spectrum of H-gas without S-Free compared to the spectrum of ambient air. The reduced peak height of the RIP in the spectrum of natural gas is caused by the different compounds of natural gas visible in the peaks next to the RIP at higher drift times. There is no peak in the drift time area of interest for the determination of S-Free, thus the concentration for S-Free would be calculated to be 0 mg m−3.
 | ||
| Fig. 5 Spectrum of ambient air and of natural gas (H-gas) without S-Free. | ||
Due to the fact that the proton affinity of the organic substances increases with the number of carbon atoms, the effectiveness of the ionisation of the odorant can be influenced due to the quality differences of L- and H-gas matrices, and therefore influence the exact quantification of S-Free.
The calibration curves obtained in the different natural gas matrices are displayed in Fig. 6. The error of the threefold measurements is just ±0.5 mg m−3 over all measurement points, which is evidence of a good reproducibility.
 | ||
| Fig. 6 Calibration curves of S-Free in L- and H-gases. | ||
Comparing the calibration curves in Fig. 6, the curve of L-gas is located below the H-gas curve. This result is a consequence of the theoretical thesis mentioned above, that the effectiveness of the ionisation is lower in L-gas due to the difference in the homogeneity of the natural gas matrix. Considering this influence, the calibration of the devices has to be investigated with the same type of natural gas as used in the gas network, which is subject to further investigations.
The validation of the μIMS in situ measurements were performed using two IMS systems and a portable gas chromatograph (type: μGC 4900 made by Varian) at six measuring points at a gas net located in eastern Germany. The quantification of S-Free was carried out with intervals of 15 minutes using the various instruments at each control point to avoid possible local concentration changes.
The comparison of the results of the different IMS systems and the gas chromatograph are displayed in Fig. 7 and show that a difference of less than 2 mg m−3 could be observed while inspecting various odorant concentrations at several measuring points.
 | ||
| Fig. 7 Results of in situ measurements at a gas net in eastern Germany. | ||
This rather good compliance between the three analytical instruments achieved during measurements at different gas stations gives evidence of the fact that the IMS technology and the product development of the μIMS as a handheld analytical device for the short-time quantification of Gasodor™ S-Free™ works with high accuracy and reproducibility.
Conclusion
A handheld instrument based on the principle of an ion mobility spectrometry was developed for the short-time determination of the concentration of sulfur-free odorant direct at the gas pipe. The results of the experiments show that IMS without pre-separation by GC-columns could be successfully used to determine the odorant level of sulfur-free odorants, such as Gasodor™ S-Free™, in natural gas within the range 4 mg m−3 to 23 mg m−3, with an accuracy of ±2 mg m−3. The quality differences of the matrix of the natural gas in the case of L- and H-gases may influence the analysis, which has to be taken into account when considering the calibration of the devices. With the help of in situ measurements, the applicability of μIMS for the on-site control of the odorant level could be successfully realized.Acknowledgements
The support of the E. ON Ruhrgas and the excellent cooperation with the DBI GUT is gratefully acknowledged.References
- H. Kaesler, T. Anderbrügge, Messtechnische Erkenntnisse – Erfahrungsaustausch Einsatz des schwefelfreien Odoriermittels Gasodor S-Free, 2004, private information.
- F. Schmeer, R. Reimert and H. Kaesler, Schwefelfreie Odorierung-Erfahrungsberichte, GWF, Gas/Erdgas, 2005, 146(4), 219–226 Search PubMed.
- C. Schunk, M. Bernhardt and R. Reimert, Schwefelfreies Odorierungsmittel—Steigerung der Umweltfreundlichkeit unter Wahrung des Sicherheitsniveaus, GWF, Gas/Erdgas, 1999, 140(10), 716–721 Search PubMed.
- K. Kröger, M. Bernhard and R. Reimert, Ergebnisse eines Feldversuchs mit dem schwefelfreen Odoriermittel “Gasodor S-FREE”, GWF, Gas/Erdgas, 2001, 142, 779–784 Search PubMed.
- DIN 51855 part 7: Bestimmung des Gehaltes an Schwefelverbindungen, 1986 Search PubMed.
- M. Goschin, H. Kuhrmann, T. Pockrandt and R. Rawe, Erfahrungen im Umgang mit Odoriermittelgemischen, GWF, Gas/Erdgas, 2003, 144(1), 44–51 Search PubMed.
- Gasodor S-Free-Das erste schwefelfreie Odoriermittel für Erdgas, Information from Symrise, June 2004.
- Sicherheitsdatenblatt 421089 Gasodor™ S-Free™ D61838, Information from Symrise, August 2003.
- DVGW certificate DG-5194BN0492, November 2002.
- W. Staudinger, S-Free Schwefelfreie Odorierung—Übernahme in das DVGW-Regelwerk—Erste Erfahrungen, GWF Gas/Erdgas, 2003, 144(10), 577–580 Search PubMed.
- K. Kröger, Bestimmung des Gehaltes von Gasodor™ S-Free™ in Erdgas mit dem Micro-Gaschromatographen. Workshop Messtechnik zur Bestimmung der Konzentration des schwefelfreien Odoriermittels Gasodor™ S-Free™ in Erdgas, Karlsruhe, 29 July 2004.
- F. Schmeer, Einführung, Workshop Messtechnik zur Bestimmung der Konzentration des schwefelfreien Odoriermittels Gasodor S-Free in Erdgas, Karlsruhe, 29 July 2004.
- Dräger-Röhrchen Messsysteme—Information of Dräger Safety, 31 December 2003.
- J. I. Baumbach and G. A. Eiceman, Ion mobility spectrometry: Arriving on-site and moving beyond a low profile, Appl. Spectrosc., 1999, 53/9, 338A–355A CrossRef CAS.
- G. A. Eiceman and Z. Karpas, Ion Mobility Spectrometry, CRC Press, Boca Raton, FL, USA, 2005 Search PubMed.
- J. I. Baumbach, Process analysis using ion mobility spectrometry, Anal. Bioanal. Chem., 2006, 384, 1059–1070 CrossRef CAS.
- J. Stach and J. I. Baumbach, Ion mobility spectrometry—basic elements and applications, Int. J. Ion Mobility Spectrom., 2002, 5(1), 1–21 Search PubMed.
- S. Sielemann, J. I. Baumbach, H. Schmidt and P. Pilzecker, Detection of alcohols using UV-ion mobility spectrometers, Anal. Chim. Acta, 2001, 431, 293–301 CrossRef CAS.
- J. I. Baumbach, P. Pilzecker and E. Trindade, Monitoring of circuit breakers using ion mobility spectrometry for sensing of SF6-decomposition, Int. J. Ion Mobility Spectrom., 1999, 2, 35–39 Search PubMed.
| This journal is © The Royal Society of Chemistry 2007 |