Thioester hydrolysis reactivity of zinc hydroxide complexes: investigating reactivity relevant to glyoxalase II enzymes†
Lisa M. Berreau*a, Amrita Sahaa and Atta M. Arifb
aDepartment of Chemistry and Biochemistry, Utah State University, 0300 Old Main Hill, Logan, UT 84322-0300, USA. E-mail: berreau@cc.usu.edu; Fax: +1 435-797-3390; Tel: +1 435-797-1625
bDepartment of Chemistry, University of Utah, 315 S. 1400 E., Salt Lake City, UT 84112-0850, USA. E-mail: arif@chemistry.utah.edu
First published on 23rd November 2005
Abstract
A recently reported binuclear zinc hydroxide complex [(L1Zn2)(µ-OH)](ClO4)2 (1, L1 = 2,6-bis[(bis(2-pyridylmethyl)amino)methyl]-4-methylphenolate monoanion) containing a single bridging hydroxide was examined for thioester hydrolysis reactivity. Treatment of 1 with hydroxyphenylthioacetic acid S-methyl ester in dry CD3CN results in no reaction after ∼65 h at 45(1) °C. Binuclear zinc hydroxide complexes of the N-methyl-N-((6-neopentylamino-2-pyridyl)methyl)-N-((2-pyridyl)methyl)amine (L2) and N-methyl-N-((6-neopentylamino-2-pyridyl)methyl)-N-((2-pyridyl)ethyl)amine (L3) chelate ligands were prepared by treatment of each ligand with molar equivalent amounts of Zn(ClO4)2·6H2O and KOH in methanol. These complexes, [(L2Zn)2(µ-OH)2](ClO4)2 (2) and [(L3Zn)2(µ-OH)2](ClO4)2 (3), which have been structurally characterized by X-ray crystallography, behave as 1 : 1 electrolytes in acetonitrile, indicating that the binuclear cations dissociate into monomeric zinc hydroxide species in solution. Treatment of 2 or 3 with one equivalent of hydroxyphenylthioacetic acid S-methyl ester per zinc center in acetonitrile results in the formation of a zinc α-hydroxycarboxylate complex, [(L2)Zn(O2CCH(OH)Ph)]ClO4·1.5H2O (4) or [(L3)Zn(O2CCH(OH)Ph)]ClO4·1.5H2O (5), and CH3SH. These reactions, to our knowledge, are the first reported examples of thioester hydrolysis mediated by zinc hydroxide complexes. The results of this study suggest that a terminal Zn–OH moiety may be required for hydrolysis reactivity with a thioester substrate.
Introduction
The glyoxalase system, which is comprised of two metalloenzymes, glyoxalase I (GLX1) and glyoxalase II (GLX2) and is found in several organisms including humans, catalyzes the conversion of 2-oxoaldehydes to 2-hydroxycarboxylic acids.1 An important substrate for this system is methyl glyoxal, CH3C(O)CHO, a reactive small molecule that is produced as a byproduct of lipid and carbohydrate metabolism.2,3 Methyl glyoxal is toxic and mutagenic due to the reactivity of the aldehyde group with guanine residues of DNA and amino acid nucleophiles, such as the amino groups of arginine and lysine, and the thiolate moiety of cysteine.4,5 As shown in Scheme 1, GLX1 catalyzes the isomerization of a hemithioacetal to yield a 2-hydroxythioester. GLX2 catalyzes the hydrolysis of this thioester to produce a 2-hydroxy acid (e.g. lactic acid) and free glutathione.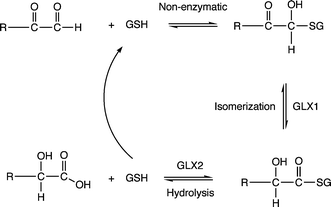 | ||
| Scheme 1 Reactions catalyzed by GLX1 and GLX2. GSH is reduced glutathione. | ||
Crystal structures of human GLX2 derived from two different crystallization conditions have been reported.6 Based on sequence homology, GLX2 has been identified as a member of the metallo-β-lactamase superfamily of hydrolases7,8 and contains a binuclear metal cluster within the active site. Metal analysis of the protein used for X-ray crystallographic studies indicated the presence of ∼1.5 moles of zinc and ∼0.7 moles of iron per protein. Despite this variable metal ion content, the structure of human GLX2 was described as having a binuclear zinc active site cluster, a description that was based in part on comparison of human GLX2 to the analogous enzyme isolated from A. thaliana, which was initially described as a zinc enzyme.6,9 Overall, the results of the crystallographic studies indicate that the two metal centers in GLX2 are separated by ∼3.3–3.5 Å, depending on crystallization conditions, and are bridged by a water/hydroxo moiety and a single oxygen atom from Asp134 (Fig. 1). The remaining zinc ligands are nitrogen atom donors from histidine ligands, and one monodentate aspartate donor coordinated to the M2 ion.
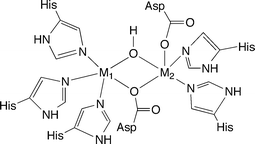 | ||
| Fig. 1 Structural features of the binuclear metal cluster in the active site of human GLX2. The description of the X-ray structure has M1/M2 = Zn(II).6 | ||
The metal coordination environment in human GLX2 is influenced by the presence of anions and substrate/inhibitor-type molecules. For example, a crystallographic data set collected on human GLX2 crystals grown from a solution containing acetate and cacodylate ((CH3)2AsO2−) ions revealed an asymmetric unit comprised of two binuclear metal sites, both of which contain a coordinated bridging µ-1,2-O2As(CH3)2 anion. In this case, the metal centers each exhibit an overall coordination number of six and a distorted octahedral geometry. A second crystallographic data set, obtained from crystals soaked in a solution containing the slow substrate S-(N-hydroxy-N-bromophenylcarbamoyl)glutathione (HBPC-GSH), revealed a HBPC-GSH substrate molecule bound near one binuclear metal site of the asymmetric unit, whereas the product GSH (M2⋯S(thiol) ∼2.9 Å) was found located near the other binuclear metal site. In the HBPC-GSH bound site of the asymmetric unit, the thioester carbonyl oxygen is positioned near M1 (M1⋯O(thioester) ∼3.3 Å) and the HBPC-GSH sulfur atom is ∼3.3 Å from M2. In this structural arrangement, the bridging water/hydroxo moiety is ∼2.9 Å from the carbonyl carbon of the thioester substrate. The α-hydroxyl group of HBPC-GSH does not interact with the binuclear metal center or via hydrogen bonding with the protein.
Based on these X-ray crystallographic studies, a mechanism for thioester hydrolysis by zinc-containing human GLX2 has been proposed (Scheme 2).6 This pathway involves attack of the bridging hydroxide moiety on the thioester carbonyl group, with the Zn1 center being involved in stabilization of the transition state. It has also been suggested that Zn2 may stabilize the glutathione thiolate moiety formed upon C–S bond cleavage.
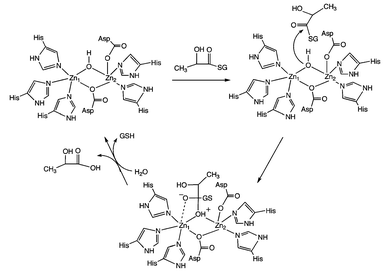 | ||
| Scheme 2 Proposed mechanism for zinc-containing human GLX2.6 | ||
In the past few years, extensive studies have been performed on the cytoplasmic GLX2 enzyme obtained from Arabidopsis thaliana, which has more than 50% sequence identity to human GLX2, and has been cloned and overexpressed in E. coli.10–12 Important findings of these studies include that: (1) GLX2 from A. thaliana can utilize either zinc, iron or manganese as the active site metal ion in vivo, (2) metal ion binding is influenced by both primary and secondary shell metal ligands, and (3) metal ion content for the enzyme depends on growth conditions. In addition, combined EPR, EXAFS, mass spectrometry and metal analysis studies indicate that metal coordination in GLX2 occurs in a non site-specific fashion and exclusively as dimetal centers that may be homogeneous or mixed-metal combinations (e.g. Fe(III)Fe(II), Fe(II)Fe(II), Mn(II)Mn(II), Fe(II)Mn(II), Fe(III)Zn(II), Mn(II)Zn(II), Fe(II)Zn(II) and Zn(II)Zn(II)).11,12 Notably, similar levels of enzymatic activity are observed irrespective of the metal ion content. Finally, as no report exists to date in the literature of the metal ion content of a GLX2 enzyme purified from a native source, the specific in vivo metal ion content of GLX2 enzymes from various sources remains unclear.
Multiple aspects of the proposed chemistry of GLX2 enzymes remain to be fully investigated. For example, in a recent mechanistic proposal for S-D-lactolylglutathione hydrolysis by A. thaliana GLX2, Makaroff and coworkers proposed that interaction of the substrate with the active site metal centers induces the bridging hydroxide moiety to become terminal on M2 prior to attack on the substrate.10 This is consistent with proposed mechanisms for other binuclear hydrolases, such as leucine aminopeptidase,13 but contrasts with the previously proposed mechanistic pathway for human GLX2 (Scheme 2).6 Furthermore, it has been reported that product release occurs in an ordered fashion, with protonation and release first of lactic acid, followed by protonation of a metal-stabilized gluthione thiolate moiety and release of the free thiol.10 This ordering indicates that the more basic thiolate is stabilized, perhaps through metal coordination. These issues, along with the impact of variable metal ion contents on the reaction pathway of GLX2 enzymes, remain to be fully examined.
In the work presented herein, we outline studies of the thioester cleavage reactivity of a series of zinc hydroxide complexes. While the first complex mimics some of the structural features of the active site binuclear metal center identified in the crystal structure of human GLX2, including the presence of a single bridging hydroxide ligand, it has been found to unreactive toward a model thioester substrate. A second type of binuclear complex, capable of forming mononuclear Zn–OH units in solution, does mediate the hydrolysis of a thioester substrate to give a zinc 2-hydroxyacid complex and free thiol.
Results and discussion
A binuclear zinc hydroxide complex of the 2,6-bis[(bis(2-pyridylmethyl)amino)methyl]-4-methylphenol (L1H) ligand,14 [(L1Zn2)(µ-OH)](ClO4)2 (1, Fig. 2), was recently reported.15 We independently prepared this complex via treatment of L1H with Zn(ClO4)2·6H2O in the presence of two equivalents of (CH3)4NOH·5H2O, which yielded 1 in 82% yield following recrystallization from acetonitrile/diethyl ether (see supplementary information for details). Complex 1 was characterized by X-ray crystallography, 1H and 13C NMR, IR spectroscopy, mass spectrometry, and elemental analysis.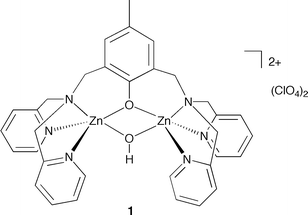 | ||
| Fig. 2 Structural drawing of 1. | ||
A crystalline form of 1, 1·3CH3CN, was obtained from acetonitrile–diethyl ether. This form belongs to the monoclinic crystal system and the P21/c space group. In this structure, there are three molecules of acetonitrile per cation. This crystalline form differs from a previously reported crystalline sample of 1 (1·CH3CN, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) wherein there is one acetontrile molecule per cation.15 We note that the overall quality of the structure of 1·3CH3CN (R1/wR2 = 0.0488/0.0854) is significantly better than that reported for 1·CH3CN (R1/wR2 = 0.095/0.199).15 That being said, the metrical parameters for the cationic component of 1·3CH3CN are similar to those previously reported for 1·CH3CN.15 For example, in each structure the zinc centers exhibit a distorted trigonal bipyramidal geometry.16 In addition, in both structures, the Zn–O(H) distances (1·3CH3CN: 1.984(2) and 1.985(2) Å; 1·CH3CN: 1.971(3) and 1.943(3) Å) are slightly shorter than the Zn–O(phenolate) distances (1·3CH3CN: 2.0260(18) and 2.0511(19) Å; 1·CH3CN: 2.033(3) and 2.030(3) Å). The cationic portion of the X-ray structure of 1·3CH3CN is shown in Fig. S1 (ESI†). Details of the X-ray data collection and refinement for 1·3CH3CN are given in Table S1 (ESI†). Selected bond distances and angles are given in Table S2 (ESI†).
) wherein there is one acetontrile molecule per cation.15 We note that the overall quality of the structure of 1·3CH3CN (R1/wR2 = 0.0488/0.0854) is significantly better than that reported for 1·CH3CN (R1/wR2 = 0.095/0.199).15 That being said, the metrical parameters for the cationic component of 1·3CH3CN are similar to those previously reported for 1·CH3CN.15 For example, in each structure the zinc centers exhibit a distorted trigonal bipyramidal geometry.16 In addition, in both structures, the Zn–O(H) distances (1·3CH3CN: 1.984(2) and 1.985(2) Å; 1·CH3CN: 1.971(3) and 1.943(3) Å) are slightly shorter than the Zn–O(phenolate) distances (1·3CH3CN: 2.0260(18) and 2.0511(19) Å; 1·CH3CN: 2.033(3) and 2.030(3) Å). The cationic portion of the X-ray structure of 1·3CH3CN is shown in Fig. S1 (ESI†). Details of the X-ray data collection and refinement for 1·3CH3CN are given in Table S1 (ESI†). Selected bond distances and angles are given in Table S2 (ESI†).
Important differences are apparent upon comparison of the structural features of the binuclear core of 1·3CH3CN with those of the active site metal cluster in GLX2.6 For example, the metal ions in the protein exhibit a different overall geometry, having an approximately square pyramidal coordination environment in the absence of substrate. In addition, the M1–O(Asp) interaction (2.2–2.6 Å) is much weaker than the M2–O(Asp) interaction (2.0–2.2 Å). This asymmetry within the core, wherein the aspartate residue between the metal centers is essentially monodentate on M2, is interesting and may be important in regard to reactivity of the bridging M–OH moiety. Specifically, in a proposed mechanism for thioester hydrolysis catalyzed by the GLX2 enzyme from Arabidopsis thaliana, Makaroff and coworkers have suggested that upon interaction of the thioester carbonyl oxygen with M1 the bridging hydroxide moves to a terminal position on M2.10 In comparison, the core in 1·3CH3CN is symmetric, with identical Zn–O(H) distances that are on the short end of the range found in GLX2 (1.9–2.2 Å).6 The M⋯M distance in the enzyme (3.3–3.5 Å) is significantly longer than that found in 1·3CH3CN (3.05 Å).
The acetonitrile solvate molecules found in the X-ray structure of 1·3CH3CN are lost upon drying of the bulk sample, 1H and 13C NMR studies of 1 in CD3CN give the expected features, albeit a 1H NMR resonance could not be identified for the bridging hydroxyl proton. Evidence for the hydroxide moiety is found in the solid state IR spectrum of 1, wherein a broad O–H stretch is found at 3430 cm−1. The broad nature of this feature may be due to hydrogen bonding involving a perchlorate anion. In this regard, in the X-ray structure of 1·3CH3CN, weak hydrogen bonding interactions are found between the hydroxyl proton and two perchlorate oxygen atoms (heteroatom distances: O(2)⋯O(9′) 3.024(8) Å, O(2)⋯O(10) 2.980(6) Å).
Reactivity studies of 1 with a thioester
The model thioester substrate hydroxyphenylthioacetic acid S-methyl ester was prepared in two steps according to literature procedures as shown in Scheme 3.17,181H NMR analysis of the hemithioacetal product of reaction A (Scheme 3) revealed diagnostic –CH– and –SCH3 resonances at 6.23 and 2.10 ppm, respectively, in CD3CN. For the thioester product from reaction B (Scheme 3) these resonances are found at 5.23 and 2.21 ppm, respectively.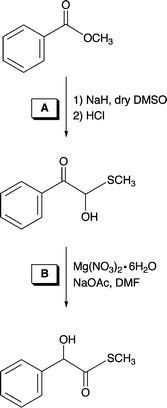 | ||
| Scheme 3 Synthetic route used for the preparation of the thioester model substrate hydroxyphenylthioacteic acid S-methyl ester.17,18 | ||
Treatment of 1 with hydroxyphenylthioacetic acid S-methyl ester in CD3CN at 45(1) °C for ∼65 h resulted in no reaction as determined by 1H NMR. Analysis of a space-filling model of the cationic portion of 1·3CH3CN (Fig. S2, ESI†) revealed that the zinc centers and bridging hydroxide moiety in this complex are exposed to solvent and thus accessible to interactions with the thioester molecule. We hypothesize that the lack of reactivity in this system is due to the poor nucleophilic character of the bridging hydroxide. However, weak binding of the thioester substrate to the zinc cation may also be an issue. In addition, we suspect that because of the symmetrical nature of the coordination environment imparted by the L1 chelate ligand, it is unlikely that the bridging hydroxide moiety will shift to a terminal position.
Synthesis and characterization of new Zn2(µ-OH)2 complexes
As a portion of our study, we have also investigated thioester cleavage chemistry involving new binuclear zinc hydroxide complexes supported by N3-donor type ligands. As shown in Schemes 4 and 5, two new N3-donor ligands (L2 and L3), each having one internal hydrogen bond donor, have been constructed. Treatment of N-methyl-N-(2-pyridylmethyl)amine (Array Biopharma) with an equimolar amount of 2-pivaloylamido-6-bromomethylpyridine,19 followed by reduction of the amide-containing ligand precursor using LiAlH4, gave L2 in modest yield (Scheme 4). The L3 ligand was also prepared via LiAlH4 reduction of an amide-appended precursor (Scheme 5).20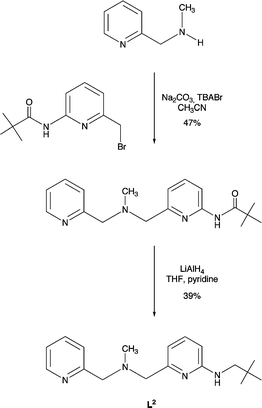 | ||
| Scheme 4 Synthesis of L2 ligand. | ||
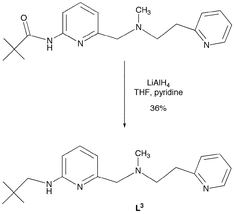 | ||
| Scheme 5 Synthesis of L3 ligand from amide-appended precursor.20 | ||
Treatment of L2 or L3 with equimolar amounts of Zn(ClO4)2·6H2O and KOH in CH3OH, followed by recrystallization from CH3CN–Et2O (L2) or CH3CN–CH2Cl2–Et2O (L3), yielded crystals suitable for single-crystal X-ray crystallography. The hydroxide complexes, [(L2Zn)2(µ-OH)2](ClO4)2 (2) and [(L3Zn)2(µ-OH)2](ClO4)2·0.5CH3CN (3), have also been characterized by 1H and 13C NMR, FTIR, and elemental analysis.
The cationic portions of 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN are shown in Fig. 3. Information concerning the X-ray data acquisition and refinement is given in Table 1. Selected bond distances and angles for 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN are presented in Table 2.
| 2·CH3CN | 3·0.5CH2Cl2·0.5CH3CN | |
|---|---|---|
| a Radiation used: Mo-Kα (λ = 0.71073 Å).b R1 = ∑‖Fo| − |Fc‖/∑|Fo|; wR2 = [∑[w(Fo2-Fc2)2]/[∑(Fo2)2]]1/2 where w = 1/[σ2(Fo2) + (aP)2 + bP]. | ||
| Empirical formula | C40H60Cl2N10O10Zn2 | C39.50H60.50Cl3N8.50O10Zn2 |
| M | 1042.62 | 1051.55 |
| Crystal system | Monoclinic | Tetragonal |
| Space group | C2/c | P4322 |
| a/Å | 16.5217(3) | 11.4472(7) |
| b/Å | 16.3421(2) | 11.4472(7) |
| c/Å | 19.4004(4) | 37.628(2) |
| β/° | 112.8344(8) | 90 |
| V/Å3 | 4827.58(15) | 4930.8(5) |
| Z | 4 | 4 |
| Dc/Mg m−3 | 1.435 | 1.417 |
| T/K | 150(1) | 150(1) |
| Color | Colorless | Colorless |
| Crystal size/mm | 0.25 × 0.25 × 0.23 | 0.33 × 0.28 × 0.25 |
| Diffractometera | Nonius KappaCCD | Nonius KappaCCD |
| µ/mm−1 | 1.168 | 1.196 |
| 2θmax/Å | 54.94 | 55.00 |
| Completeness to θ = 27.94° (%) | 99.7 | 96.7 |
| Reflections collected | 9712 | 9994 |
| Independent reflections | 5522 | 5428 |
| Rint | 0.0185 | 0.0433 |
| Variable parameters | 330 | 300 |
| R1/wR2b | 0.0540/0.1267 | 0.0556/0.1034 |
| Goodness-of-fit (F2) | 1.220 | 1.100 |
| Δρmax min/e Å−3 | 0.490/−0.567 | 0.482/−0.602 |
| 2·CH3CN | |||
| Zn(1)–O(1) | 1.983(3) | Zn(1)–N(3) | 2.105(3) |
| Zn(1)–O(1)#1 | 2.038(3) | Zn(1)–N(4) | 2.225(3) |
| Zn(1)–N(2) | 2.079(3) | Zn(1)⋯Zn(1)#1 | 2.9838(8) |
| O(1)–Zn(1)–O(1)#1 | 80.32(12) | N(2)–Zn(1)–N(4) | 119.83(12) |
| O(1)–Zn(1)–N(2) | 126.53(11) | O(1)–Zn(1)–N(3) | 99.75(11) |
| O(1)#1–Zn(1)–N(2) | 101.33(11) | O(1)#1–Zn(1)–N(3) | 178.39(11) |
| O(1)–Zn(1)–N(4) | 111.88(12) | N(2)–Zn(1)–N(3) | 79.95(11) |
| O(1)#1–Zn(1)–N(4) | 101.28(11) | N(4)–Zn(1)–N(3) | 77.20(12) |
| 3·0.5CH2Cl2·0.5CH3CN | |||
|---|---|---|---|
| a Estimated standard deviations indicated in parentheses. | |||
| Zn(1)–O(1) | 2.070(3) | Zn(1)–N(3) | 2.243(4) |
| Zn(1)–O(1)#1 | 1.963(3) | Zn(1)–N(4) | 2.099(4) |
| Zn(1)–N(2) | 2.079(4) | Zn(1)⋯Zn(1)#1 | 2.9871(10) |
| O(1)–Zn(1)–O(1)#1 | 79.10(17) | N(2)–Zn(1)–N(4) | 113.77(15) |
| O(1)–Zn(1)–N(2) | 100.20(14) | O(1)–Zn(1)–N(3) | 178.12(16) |
| O(1)#1–Zn(1)–N(2) | 125.78(14) | O(1)#1–Zn(1)–N(3) | 99.26(15) |
| O(1)–Zn(1)–N(4) | 95.50(15) | N(2)–Zn(1)–N(3) | 79.99(15) |
| O(1)#1–Zn(1)–N(4) | 120.30(15) | N(4)–Zn(1)–N(3) | 86.11(16) |
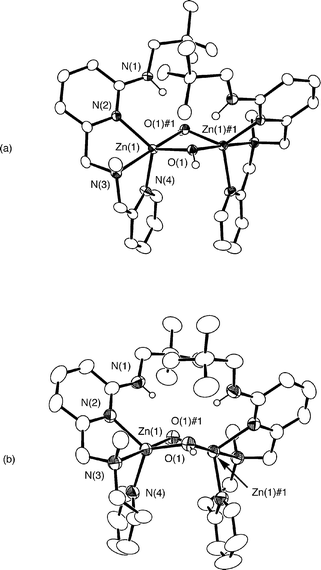 | ||
| Fig. 3 ORTEP drawings of the cationic portions of 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN. All hydrogen atoms, except the hydroxyl and secondary amine protons, have been omitted for clarity. Ellipsoids are drawn at the 50% probability level for 2·CH3CN and at the 35% probability level for 3·0.5CH2Cl2·0.5CH3CN. | ||
The cationic portions of 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN are comprised of a binuclear Zn2(µ-OH)2 core supported at each zinc center by a N3-donor ligand. The core has a hinge-type distortion wherein each bridging hydroxide oxygen atom accepts a hydrogen bond from the secondary amine group of the supporting chelate ligand. These secondary interactions may be classified as moderate hydrogen bonds on the basis of the heteroatom bond distances and angles (2·CH3CN: N(1)⋯O(1)#1 2.852(4) Å, N(1)–H(1N)⋯O(1)#1 164(4)°; 3·0.5CH2Cl2·0.5CH3CN: N(1)⋯O(1) 2.864(6) Å, N(1)–H(1N)⋯O(1) 158(4)°).21 Each hydroxyl proton is also involved in a hydrogen bonding interaction with an oxygen atom of a perchlorate anion. The hinge-type core found in 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN is similar to that found in [(bmnpaCd)2(µ-OH)2](ClO4)2 (bmnpa = N,N-bis-2-(methylthio)ethyl-N-((6-neopentylamino-2-pyridyl)methyl)amine) (Fig. 4(a)).22 In this binuclear cadmium hydroxide complex, the positioning of both neopentyl amino substituents on one side of the Cd2(µ-OH)2 core results in CH/π interactions between the neopentyl hydrogens of one chelate ligand and the pyridyl ring of the other chelate ligand.23,24 These interactions are characterized by methyl or methylene carbon/arene centroid distances of 3.15–3.84 Å. In the solid-state structures of 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN, the shortest methyl carbon/arene centroid distances are 4.06 and 4.34 Å, respectively. As these distances exceed the sum of the van der Waals radii of a methyl group and a Csp2 unit (3.7 Å),24 no CH/π interactions are present in the solid-state structures of 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN.
2 and (b) [(bmnpa/benpaZn)2(µ-OH)2](ClO4)2.22,25,26](/image/article/2006/DT/b512515d/b512515d-f4.gif) | ||
| Fig. 4 Structural drawings of (a) [(bmnpaCd)2(µ-OH)2](ClO4)2 and (b) [(bmnpa/benpaZn)2(µ-OH)2](ClO4)2.22,25,26 | ||
The symmetry-related zinc centers in 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN exhibit a distorted trigonal bipyramidal geometry (τ = 0.86, 2·CH3CN; τ = 0.87, 3·0.5CH2Cl2·0.5CH3CN) with two pyridyl nitrogens and a hydroxide oxygen atom occupying equatorial positions.16 The Zn⋯Zn distances (2.9838(8), 2.9871(10) Å) are comparable to the Zn⋯Zn distances in the binuclear zinc hydroxide derivatives [(bmnpaZn)2(µ-OH)2](ClO4)2 (2.98 Å)25 and [(benpaZn)2(µ-OH)2](ClO4)2 (2.97 Å; benpa = N,N-bis-2-(ethylthio)ethyl-N-((6-neopentylamino-2-pyridyl)methyl)amine) (Fig. 4(b)).26 Two slightly different Zn–O(H) distances are found within the cationic portions of 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN. These distances (Table 2) are within the range (1.9–2.3 Å) previously reported for bridging hydroxide ligands in multinuclear zinc complexes.25 Similar to the core structures of [(bmnpaZn)2(µ-OH)2](ClO4)2 and [(benpaZn)2(µ-OH)2](ClO4)2, in 2·CH3CN and 3·0.5CH2Cl2·0.5CH3CN, the longer Zn–O(H) distance involves the oxygen atom that accepts a hydrogen bond from the supporting chelate ligand on the same zinc center.
In CH3CN solution, analytically pure samples of 2 and 3 behave as 1 : 1 electrolytes, thus indicating a breakup of the binuclear cations in these complexes into [(L2/L3)Zn–OH]ClO4 fragments. This is evident from the Onsager slopes exhibited by these complexes in CH3CN solution, as shown in Fig. 5, which are similar to that of the 1 : 1 electrolyte standard Me4NClO4. For comparison, the conductance data for [(bmnpaZn)2(µ-OH)2](ClO4)225 is also shown in Fig. 5. The higher slope value for this complex indicates the presence of a 1 : 2 electrolyte.27
2;25 (×), 2 (□), 3 (○) and Me4NClO4 (◇).](/image/article/2006/DT/b512515d/b512515d-f5.gif) | ||
| Fig. 5 Onsager plots for [(bmnpaZn)2(µ-OH)2](ClO4)2;25 (×), 2 (□), 3 (○) and Me4NClO4 (◇). | ||
The 1H NMR features of analytically pure 2 in dry CD3CN at ambient temperature are sharp and indicative of the presence of a major and minor species at ambient temperature (Fig. S3(a), ESI†). For the major species, a sharp N–H resonance is present at 9.18 ppm. This signal appears as a triplet (J = 5.2 Hz) due to coupling with the adjacent methylene protons in the neopentyl portion of the L2 ligand. This observed coupling, along with the deshielding of the N–H proton resonance, provides evidence for the involvement of the secondary amine N–H in a hydrogen bonding interaction.21 Based on the solid state X-ray structure of 2·CH3CN and the above-mentioned conductance experiments, we propose that this hydrogen bonding interaction involves the Zn–OH moiety of a mononuclear [(L2)Zn–OH]ClO4 species in solution. Additionally, the protons of the benzylic positions of the major species are diastereotopic, appearing as four doublets in the chemical shift range of 3.4–4.5 ppm. The neopentyl methylene protons of the major species appear as a doublet of doublets. The magnitude of the coupling constants in this signal indicates that these protons are diastereotopic and each coupled to the secondary amine N–H proton. The minor species present in dry CD3CN solutions of 2 has a tert-butyl methyl signal at 0.77 ppm and remains to be conclusively identified. However, based on the fact that a binuclear cation is present in the solid state structure of 2·CH3CN, we propose that the minor species may be a binuclear form of the complex.
The 1H NMR features of analytically pure 3 in dry CD3CN (Fig. S3(b), ESI†) at ambient temperature also indicate the presence of two species, albeit the ratio is different than that found for 2. The major and minor species for 3 have secondary amine N–H resonances at 9.24 and 9.06 ppm, respectively. Integration of these signals provides a ratio of ∼2 : 1 for the major vs. minor species. The tert-butyl methyl resonance for the minor species (0.85 ppm) is again upfield of that of the major species (0.90 ppm). Speculating that these two species may be mono- and binuclear forms of 3, we investigated CD3CN solutions of 3 using 1H NMR at variable temperatures and concentrations. Both heating and cooling produced peak broadening that prevented careful integration of the individual resonances. Examination of the 1H NMR features of 3 in CD3CN over a ten-fold concentration range (1.8 × 10−2–1.8 × 10−3 M) produced a subtle change (∼6%) in the ratio of the integrated intensities of the tert-butyl resonances of the two species. However, as these resonances overlap at the baseline, we are hesitant to assign significance to this observed change. The conductance experiments for 3 (Fig. 5) were performed over a concentration range of 4.9 × 10−3–1.3 × 10−4 M. As the results of these measurements indicate the presence of a 1 : 1 electrolyte, we propose that the major species present in acetonitrile solutions of 3 is a mononuclear [(L3)Zn–OH]ClO4 species and that this species becomes more dominant as the concentration decreases.
Thioester hydrolysis reactivity of 2 and 3
Treatment of 2 or 3 with one equivalent of hydroxyphenylthioacetic acid S-methyl ester per Zn–OH moiety for 3 h at 45 °C in acetonitrile results in the clean formation of new zinc complexes (Scheme 6). The complexes, designated 4 and 5, have been isolated as powdered solids and have been characterized by elemental analysis, conductance measurements, and 1H and 13C NMR and IR spectroscopy. Attempts to generate crystalline samples of 4 and 5 suitable for X-ray crystallography have thus far failed.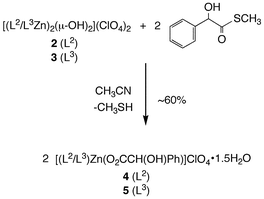 | ||
| Scheme 6 Reactivity of 2 and 3 with hydroxyphenylthioacetic acid S-methyl ester. | ||
Combustion elemental analysis data for 4 and 5 is consistent with the empirical formulas [(L2)Zn(O2CCH(OH)Ph)]ClO4·1.5H2O and [(L3)Zn(O2CCH(OH)Ph)]ClO4·1.5H2O, respectively, for these complexes. The presence of water in the powdered samples sent for elemental analysis was confirmed by evaluation of the samples by 1H NMR in dry CD3CN. Although the solid state structures of 4 and 5 are unknown, conductance measurements (Fig. 6), performed using analytically pure samples, indicate that both complexes behave as 1 : 1 electrolytes in CH3CN solution. Notably, a search of the Cambridge Crystallographic Database (CSD version 5.26 (updated May 2005)) revealed only one example of a zinc complex having a coordinated mandelate (O2CCH(OH)Ph) anion.28 This complex has two monodentate α-hydroxycarboxylate ligands.
2;25 (×), 4 (□), 5 (○) and Me4NClO4 (◇).](/image/article/2006/DT/b512515d/b512515d-f6.gif) | ||
| Fig. 6 Onsager plots for [(bmnpaZn)2(µ-OH)2](ClO4)2;25 (×), 4 (□), 5 (○) and Me4NClO4 (◇). | ||
The 1H NMR resonances of 4 and 5 in dry CD3CN are generally sharp. However, for both complexes, two broad signals are found in the range of 4–7 ppm (4: 5.45 and 6.49 ppm; 5: 4.58 and 6.82 ppm). These signals disappear in the presence of D2O and thus are likely the mandelate hydroxyl proton and secondary amine proton resonances. In the 13C NMR spectra of 4 and 5, the carboxylate carbon centers exhibit resonances at 178.7 and 179.8 ppm, respectively. The IR features of 4 and 5 related to the mandelate hydroxyl group and zinc-bound carboxylate moiety are not easily interpreted. This is because the regions of ∼3400–3200 and ∼1650–1400 cm−1 also contain vibrations associated with the supporting L2 and L3 chelate ligands. The presence of ClO4− in analytically pure samples of 4 and 5 is evident from vibrations at ∼1100 and 625 cm−1. In sum, despite the lack of X-ray crystallographic characterization for 4 or 5, analytical, conductance, and spectroscopic data are consistent with the empirical formulation of these complexes as [(L2/L3)Zn(O2CCH(OH)Ph)]ClO4·1.5H2O.
Formation of the carboxylate derivatives 4 and 5 as shown in Scheme 6 requires the release of CH3SH. We first attempted to identify CH3SH production in the reaction mixture using 1H NMR. However, as this volatile compound has a –SCH3 resonance at ∼2.0 ppm, a region that is obscured by the residual solvent resonance in CD3CN, we instead used the deuterated analog, hydroxyphenylthioacetic acid S-methyl(d3) ester. This compound, which was prepared using the procedure outlined in Scheme 4 starting from d6-DMSO, enables the use of 2H NMR as a technique to detect CD3SH production in protioacetonitrile. A reaction mixture containing 3 and hydroxyphenylthioacetic acid S-methyl(d3) ester in CH3CN initially exhibited a single 2H NMR resonance at 2.35 ppm when referenced to an internal standard of CD3NO2. However, within minutes at ambient temperature, a new 2H NMR resonance is present at 2.02 ppm. This resonance disappears if the tube is opened, indicating the formation of a volatile product.
Conclusions
In this study, we have performed an initial evaluation of the thioester cleavage reactivity of three zinc hydroxide complexes. To our knowledge, this work provides the first examples of zinc hydroxide-mediated thioester hydrolysis.29,30 Conclusions with relevance to GLX2 can be drawn from this initial study. First, as the hydroxide moiety in 1, which symmetrically bridges two structurally similar Zn(II) centers, is not sufficiently nucleophilic to react with a thioester substrate, it seems likely that a terminal or quasi-terminal metal hydroxide moiety may be required for the hydrolysis of a thioester substrate. This notion supports a proposed mechanism put forth by Makaroff and co-workers for GLX2 wherein a terminal hydroxide on M2 attacks the thioester substrate.10 Notably, we have found that zinc hydroxide complexes that dissociate into mononuclear terminal [(L)Zn–OH]ClO4 species in CH3CN solution (2 and 3) are reactive for the hydrolysis of a thioester. While kinetic studies are needed to elucidate the reactive form of the complex, in this work we have identified the reaction products as zinc mandelate complexes and CH3SH. The production of free thiol in these systems indicates that the more basic thiolate anion (vs. the carboxylate product) becomes protonated upon thioester cleavage.In terms of the possible requirement of a terminal hydroxide for thioester cleavage, we note that studies of another dinuclear metallohydrolase, purple acid phosphatase,31,32 suggest that a terminal or quasi-terminal metal hydroxide is required for the hydrolysis of a phosphate ester substrate. However, these results contrast with results of ENDOR studies of uterferrin, which suggest the involvement of a bridging hydroxide as the required nucleophile.33 Recent studies of the reactivity of dizinc complexes of varying Zn⋯Zn distance with a phosphate diester substrate revealed that hydrolytic reactivity increased with a larger Zn⋯Zn separation. This was attributed to the formation of a quasi-terminal Zn–OH species (Zn–OH⋯HO(H)–Zn).34 Overall, the question of whether a terminal or bridging hydroxide is the active nucleophile for substrate hydrolysis remains under investigation for a variety of binuclear metallohydrolases.
Finally, it is important to note that detailed mechanistic studies of thioester hydrolysis by GLX2 have been limited by the aforementioned heterogeneity of enzyme samples in terms of metal ion content for enzyme produced recombinantly in Escherichia coli.11 In light of this limitation, this is an ideal area for the use of synthetic model studies to carefully address how varying metal ion compositions influence the mechanistic pathway of thioester hydrolysis. In addition, synthetic model studies can also be used to address how the differing metal coordination environments in GLX2 influence the reactivity of a bridging hydroxide moiety. Building on the initial results presented in this study, efforts are underway in our laboratory in pursuit of these goals.
Experimental
General
All reagents and solvents were obtained from common commercial sources and were used as received unless otherwise noted. Solvents were dried according to published procedures and were distilled under N2 prior to use.35 Air sensitive procedures were performed in a MBraun Unilab glovebox under an atmosphere of purified N2. IR spectra were recorded on a Shimadzu FTIR-8400 as KBr pellets. 1H and 13C{1H} NMR spectra for characterization purposes were recorded in dry CD3CN at ambient temperature on a JEOL 270 MHz or Bruker ARX400 spectrometer. Chemical shifts (in ppm) are referenced to the residual solvent peak(s) in CHD2CN (1H: 1.94 (quintet); 13C{1H}: 1.39 (heptet) ppm). 2H NMR spectra were recorded as previously described.36 Conductance measurements were performed and analyzed using methods that have been previously described.22 Electron impact and fast atom bombardment mass spectra were obtained at the University of California, Riverside. Elemental analyses were performed by Atlantic Microlabs of Norcross, GA.The protonated form of 2,6-bis[(bis(2-pyridylmethyl)amino)methyl]-4-methylphenol (L1H), and the ligand precursor 2-pivaloylamido-6-bromomethylpyridine, were prepared according to literature procedures.14,19N-Methyl-N-(2-pyridylmethyl)amine hydrochloride was purchased from Array Biopharma and was used as received. N-methyl-N-((6-pivaloylamido-2-pyridyl)methyl)-N-(2-pyridylethyl)amine was prepared as previously described.20 The model thioester substrate hydroxyphenylthioacetic acid S-methyl ester was prepared according to literature procedures.17,18
CAUTION: Perchlorate salts of metal complexes with organic ligands are potentially explosive. Only small amounts of material should be prepared and these should be handled with great care.37
Synthesis of ligands and complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1689; δH (400 MHz, solvent CD3CN): 1.27 (9H, s), 2.22 (3H, s), 3.61 (2H, s), 3.70 (2H, s), 7.16–7.21 (1H, m), 7.25 (1H, d, J = 7.5 Hz), 7.51 (1H, d, J = 7.8 Hz), 7.70 (2H, t, J = 7.8 Hz), 7.99 (1H, d, J = 8.2 Hz), 8.20 (1H, br, N–H), 8.48 (1H, d, J = 4.2 Hz); δC (100 MHz, solvent CD3CN): 27.6, 40.5, 43.0, 64.0, 64.4, 112.8, 119.6, 123.1, 123.9, 137.4, 139.6, 150.0, 152.4, 159.3, 160.6, 178.0 (16 signals expected and observed).
O) 1689; δH (400 MHz, solvent CD3CN): 1.27 (9H, s), 2.22 (3H, s), 3.61 (2H, s), 3.70 (2H, s), 7.16–7.21 (1H, m), 7.25 (1H, d, J = 7.5 Hz), 7.51 (1H, d, J = 7.8 Hz), 7.70 (2H, t, J = 7.8 Hz), 7.99 (1H, d, J = 8.2 Hz), 8.20 (1H, br, N–H), 8.48 (1H, d, J = 4.2 Hz); δC (100 MHz, solvent CD3CN): 27.6, 40.5, 43.0, 64.0, 64.4, 112.8, 119.6, 123.1, 123.9, 137.4, 139.6, 150.0, 152.4, 159.3, 160.6, 178.0 (16 signals expected and observed).X-Ray crystallography
Crystals of 1–3 were mounted on a glass fiber using a viscous oil and then transferred to a Nonius KappaCCD diffractometer with Mo-Kα radiation (λ = 0.71073 Å) for data collection at 150(1) K. For each compound, an initial set of cell constants was obtained from 10 frames of data that were collected with an oscillation range of 1 degree frame−1 and an exposure time of 20 s frame−1. Indexing and unit cell refinement based on all observed reflections from those 10 frames indicated a monoclinic P lattice for 1·3CH3CN, a monoclinic C lattice for 2·2CH3CN, and a tetragonal P lattice for 3·0.5CH2Cl2·0.5CH3CN. Final cell constants for each complex were determined from a set of strong reflections from the actual data collection. For each data set, reflections were indexed, integrated, and corrected for Lorentz, polarization, and absorption effects using DENZO-SMN and SCALEPAC.38 The structures of 1·3CH3CN, 2·2CH3CN, and 3·0.5CH2Cl2·0.5CH3CN were solved using a combination of direct methods and heavy atom using SIR 97.For 1·3CH3CN, the hydrogen atom on the bridging hydroxide was located and refined independently using SHELXL97.39 All other hydrogen atoms for 1·3CH3CN were assigned isotropic displacement coefficients U(H) (1.2 U(C) or 1.5 U(Cmethyl), and their coordinates were allowed to ride on their respective carbons using SHELXL97.38 There is one disordered perchlorate anion in 1·3CH3CN. The O(8), O(9) and O(10) oxygen atoms, bonded to Cl(2), were each split into two fragments (O(8)/O(8′), O(9)/O(9′), O(10)/O(10′)) and were refined. This refinement led to a 60 : 40 ratio in occupancy for each atom over the two positions. The hydroxyl proton of 1·3CH3CN participates in a weak hydrogen bonding interaction with two perchlorate oxygen atoms.
For 2·2CH3CN and 3·0.5CH2Cl2·0.5CH3CN, the hydrogen atom on the bridging hydroxide and the secondary amine hydrogen were located and refined independently. All other hydrogen atoms were treated as riding atoms on their respective carbons as described for 1·3CH3CN. There is one highly disordered perchlorate anion in 2·2CH3CN. One of the two acetonitrile solvate molecules in 2·2CH3CN is also disordered. The C(20) and N(5) atoms were each split into two fragments (C(20)/C(20′), N(5)/N(5′)) and were refined. This refinement led to a 50:50 ratio in occupancy over each position for each atom. In the asymmetric unit of 3·0.5CH2Cl2·0.5CH3CN there are two partially occupied solvent molecules (0.25 CH3CN, 0.25 CH2Cl2) disordered over the same position. The methylene chloride carbon atom (C(20)) is on a two-fold position. The chloride atom position overlaps with the position of the acetonitrile sp carbon. This overlap was modeled as 0.5 Cl and 0.25 nitrile carbon from the respective solvents. Thus, the total chlorine count of 1.18 is an indication of this overlap. A DUMY atom was used to assist in calculating hydrogen atoms in ideal positions for the solvent.
CCDC reference numbers 282998–283000.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512515d
Acknowledgements
This work was supported by the National Science Foundation (CAREER Award CHE-0094066). The authors thank Ewa Szajna-Fuller and Katarzyna Rudzka for assistance with experiments and manuscript preparation.References
- P. J. Thornalley, Biochem. J., 1990, 269, 1–11 CAS.
- P. J. Thornalley, Crit. Rev. Oncol. Hematol., 1995, 20, 99–128 CrossRef CAS.
- P. J. Thornalley, Chem. Biol. Interact., 1998, 111–112, 137–151 CrossRef CAS.
- A. Papoulis, Y. al-Abed and R. Bucala, Biochemistry, 1995, 34, 648–655 CrossRef CAS.
- A. Rahman, A. Shahabuddin and S. M. Hadi, J. Biochem. Toxicol., 1990, 5, 161–166 CrossRef CAS.
- A. D. Cameron, M. Ridderström, B. Olin and B. Mannervik, Struct. Fold Des., 1999, 7, 1067–1078 Search PubMed.
- S. Melino, C. Capo, B. Dragani, A. Aceto and R. Petruzzelli, Trends. Biochem. Sci., 1998, 23, 381–382 CrossRef CAS.
- C. M. Gomes, C. Frazão, A. V. Xavier, J. Legall and M. Teixeira, Protein Sci., 2002, 11, 707–712 CrossRef CAS.
- M. W. Crowder, M. K. Maiti, L. Banovic and C. A. Makaroff, FEBS Lett., 1997, 418, 351–354 CrossRef CAS.
- T. M. Zang, D. A. Hollman, P. A. Crawford, M. W. Crowder and C. A. Makaroff, J. Biol. Chem., 2001, 276, 4788–4795 CrossRef CAS.
- O. Schilling, N. Wenzel, M. Naylor, A. Vogel, M. Crowder, C. Makaroff and W. Meyer-Klauke, Biochemistry, 2003, 42, 11777–11786 CrossRef CAS.
- N. F. Wenzel, A. L. Carenbauer, M. P. Pfiester, O. Schilling, W. Meyer-Klaucke, C. A. Makaroff and M. W. Crowder, J. Biol. Inorg. Chem., 2004, 9, 429–438 Search PubMed.
- C. Stamper, B. Bennett, T. Edwards, R. C. Holz, D. Ringe and G. Petsko, Biochemistry, 2001, 40, 7035–7046 CrossRef CAS.
- A. S. Borovik, V. Papaefthymiou, L. F. Taylor, O. P. Anderson and L. Que, Jr., J. Am. Chem. Soc., 1989, 111, 6183–6195 CrossRef CAS.
- K. Matsufuji, H. Shiraishi, Y. Miyasato, T. Shiga, M. Ohba, T. Yokoyama and H. Okawa, Bull. Chem. Soc. Jpn., 2005, 851–858 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- G. A. Russell and G. J. Mikol, J. Am. Chem. Soc., 1966, 88, 5498–5504 CrossRef.
- S. S. Hall and A. Poet, Tetrahedron Lett., 1970, 2867–2868 CrossRef CAS.
- L. M. Berreau, S. Mahapatra, J. A. Halfen, V. G. Young, Jr. and W. B. Tolman, Inorg. Chem., 1996, 35, 6339–6342 CrossRef CAS.
- A. L. Fuller, R. W. Watkins, A. M. Arif and L. M. Berreau, Inorg. Chim. Acta DOI:10.1016/j.ica.2005.09.008.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- R. A. Allred, L. H. McAlexander, A. M. Arif and L. M. Berreau, Inorg. Chem., 2002, 41, 6790–6801 CrossRef CAS.
- M. Nishio, Y. Umezawa, M. Hirota and Y. Takeuchi, Tetrahedron, 1995, 51, 8665–8701 CrossRef CAS.
- M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction: Evidence, Nature and Consequences, Wiley-VCH, New York, 1998 Search PubMed.
- L. M. Berreau, R. A. Allred, M. M. Makowska-Grzyska and A. M. Arif, Chem. Commun., 2000, 1423–1424 RSC.
- D. K. Garner, R. A. Allred, K. J. Tubbs, A. M. Arif and L. M. Berreau, Inorg. Chem., 2002, 41, 3533–3541 CrossRef CAS.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- R. Carballo, B. Covelo, E. Garcia-Martinez, E. M. Vazquez-Lopez and A. Castineiras, Appl. Organomet. Chem., 2004, 18, 201–202 CrossRef CAS.
- Examples of zinc hydroxide-mediated carboxy and phosphate ester hydrolysis are prevelant: (a) G. Parkin, Chem. Rev., 2004, 104, 699–767 CrossRef CAS; (b) R. C. diTargiani, S. Chang, M. H. Salter, Jr., R. D. Hancock and D. P. Goldberg, Inorg. Chem., 2003, 42, 5825–5836 CrossRef CAS , and references therein.
- Reactivity of a zinc methoxide complex with thioester substrates has been reported: H. Brombacher and H. Vahrenkamp, Inorg. Chem., 2004, 43, 6050–6053 Search PubMed.
- C. Belle and J.-L. Pierre, Eur. J. Inorg. Chem., 2003, 4137–4146 CrossRef CAS.
- X. Wang, R. Y. N. Ho, A. K. Whiting and L. Que, Jr., J. Am. Chem. Soc., 1999, 121, 9235–9236 CrossRef CAS.
- S. K. Smoukov, L. Quaroni, X. Wang, P. E. Doan, B. M. Hoffman and L. Que, Jr., J. Am. Chem. Soc., 2002, 124, 2595–2603 CrossRef CAS.
- B. Bauer-Siebenlist, F. Meyer, E. Farkas, D. Vidovic and S. Dechert, Chem.–Eur. J., 2005, 11, 4349–4360 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth-Heinemann, Boston, MA, 4th edn, 1996 Search PubMed.
- E. Szajna, P. Dobrowolski, A. L. Fuller, A. M. Arif and L. M. Berreau, Inorg. Chem., 2004, 43, 3988–3997 CrossRef CAS.
- W. C. Wolsey, J. Chem. Educ., 1973, 50, A335–A337 CrossRef CAS.
- Z. Otwinowski and M. Minor, Methods Enzymol., 1997, 276, 307–326 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of 1. Table S1: Crystallographic data for 1·3CH3CN. Table 2: Selected bond lengths and angles for 1·3CH3CN. Fig. S1: Cationic portion of the X-ray structure of 1·3CH3CN. Fig. S2: Space-filling model of the cationic portion of 1·3CH3CN. Fig. S3: 1H NMR spectra of analytically pure 2 and 3 in dry CD3CN. See DOI: 10.1039/b512515d |
| This journal is © The Royal Society of Chemistry 2006 |