Bisoxazolines with one and two sidearms: stereodirecting ligands for copper-catalysed asymmetric allylic oxidations of alkenes
Markus
Seitz
a,
Carmine
Capacchione
ac,
Stéphane
Bellemin-Laponnaz
b,
Hubert
Wadepohl
a,
Benjamin D.
Ward
a and
Lutz H.
Gade
*a
aAnorganisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 270, 69120, Heidelberg, Germany. E-mail: lutz.gade@uni-hd.de
bLaboratoire de Chimie Organométallique et de Catalyse, Université Louis Pasteur, 4 rue Blaise Pascal, 67000, Strasbourg, France
cDipartimento di Chimica, Università degli Studi di Salerno, via S. Allende 84081, Baronissi (SA), Italy
First published on 14th November 2005
Abstract
A series of sidearm functionalized bisoxazoline ligands has been synthesized by reaction of the monolithiated methyl{bis(oxazolinyl)}methane with the appropriate electrophiles, and tested in the copper catalyzed asymmetric allylic oxidation of cyclohexene (“Kharasch–Sosnovski” reaction). The observed enantioselectivities were higher (up to 85% ee) than for the unfunctionalized bisoxazoline (“BOX”) derivatives (ca. 60% ee). Regardless of the functional groups incorporated into the sidearm unit, the ee's obtained for the different derivatives were essentially indistinguishable. This implies that the sidearms do not interfere directly in this reaction and only play an indirect role by virtue of their steric demand. Three of the copper complexes have been characterized by X-ray diffraction, establishing a distorted octahedral coordination geometry around the copper atom in all three cases. In the elongated distorted CuN2O4 octahedra, the two nitrogen atoms of the oxazolines and one oxygen atom of each acetate ligand occupy the ‘equatorial’ positions whereas the sidearms do not interact with the metal centres.
Introduction
Bisoxazolines (“BOX”) have been established as a class of “privileged” stereodirecting ligands in asymmetric catalysis.1 Their transition metal complexes have been employed in a wide range of catalytic transformations, in particular, in Lewis acid catalyzed stereoselective reactions.2 They have provided the base for extensive research efforts in the design of new chiral ligands and may be combined with other ligating functions.3 A notable development is the coupling of an additional “sidearm” to the bridging carbon atoms of bisoxazolines.4 Tang and coworkers have found that such potentially tricoordinating ligands may provide Cu-based Lewis acid catalysts which display a significantly improved performance in comparison to the BOX-reference system.4,5 However, the role of the “sidearm” remains a matter of debate.There are three conceivable ways in which a “sidearm”, which is attached to the bridging carbon atom of the BOX-ligand may interact with the active centre of a molecular catalyst (Fig. 1). First, if it contains a ligating donor function (D) it may bind directly to the metal centre, as represented for case (a) in Fig. 1. This additional binding interaction is expected to stabilize the complex and to change the electron density of the metal. If the sidearm contains a substrate acceptor function (A), such as a hydrogen bond donating functional group, as in case (b), then an additional orientation of the catalytic substrate may be the result. Finally, the addition of the sidearm may neither lead to a direct interaction with the metal centre nor the substrate but may influence the activity and selectivity of the catalyst by nature of its steric demand [case (c)] and thus be simply due to the reduction of the active space available for the catalytic conversion.
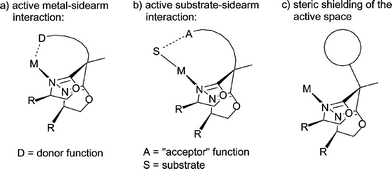 | ||
| Fig. 1 Possible roles of ligand sidearms in bisoxazolines. | ||
The introduction of the additional ligating “arm” in the podand structure of the bisoxazolines implies the loss of the twofold rotational symmetry of the system, and therefore the reduction of the number of diastereomeric intermediates and transition states in a stereoselective transformation. This complication may be in part offset by the “resymmetrization” of the system upon the introduction of two identical ligand sidearms pointing in opposite directions relative to the bisoxazoline unit. In such a system there may be rapid exchange between the interaction of the two sidearms with the metal centre or the substrate, as is exemplified in Fig. 2 for a bis(donor)-functionalized system. Here again, depending on the catalyst and the type of reaction, the role of the sidearms may also be purely steric.
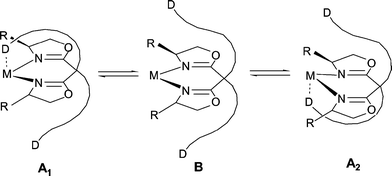 | ||
| Fig. 2 “Degenerate” interaction of the ligand sidearms in difunctionized bisoxazolines. | ||
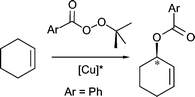
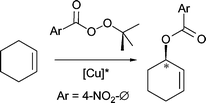
In our investigation into the catalytic utility of such additionally functionalized stereodirecting bis(oxazoline) ligands6 we chose a copper catalyzed reaction in which the copper does not primarily play the role of a Lewis acid. A preparatively useful example of a direct catalytic functionalization of hydrocarbons is the copper-catalyzed Kharasch–Sosnovsky reaction, i.e. the allylic acyloxylation of olefins.7,8 In the presence of chiral oxazoline-based ligands it has been possible to render this reaction enantioselective,9 although a truly efficient asymmetric catalyst has not as yet been found. In this work we report a series of new copper(II)-precatalysts for allylic oxidation and discuss the role that ligand sidearms may play in it.
Results and discussion
Synthesis of the ligands
All sidearm functionalized bisoxazoline ligands employed in this study were synthesized by reaction of the monolithiated methyl{bis(oxazolinyl)}methane10 with the appropriate electrophiles (Scheme 1). The lithiated starting material which was generated in situ by deprotonation in the bridgehead position of the corresponding bisoxazoline with tBuLi. Reaction of the lithium salt with a range of aroyl and acyl chlorides gave the ligands 1a–d and 2a, b while the corresponding transformation with 2-chloromethyl pyridine yielded compounds 3a, b.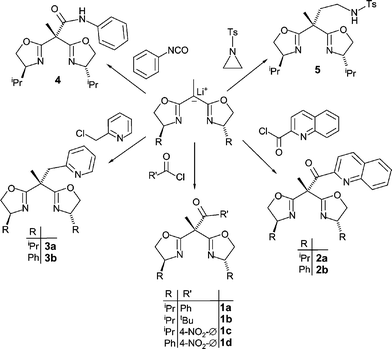 | ||
| Scheme 1 Synthesis of the bisoxazoline ligands 1–5 containing one sidearm. | ||
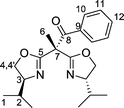
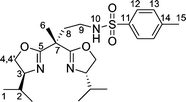
Using phenylisocyanate as the electrophilic agent gave bisoxazoline 4, containing a carboxamido sidearm, whereas ring opening of N-tosylaziridine gave rise to compound 5 possessing a sulfonylamido group in the side chain. For the synthesis of the latter it proved important to use an excess of the lithiated bisoxazoline in order to suppress the oligo- or polymerization of the N-tosylaziridine. Compound 5 is characterized by the strong ν(RSO2N)-vibrational bands in the IR spectrum at 1326 cm−1 and 1158 cm−1. After recrystallization from n-hexane single crystals of compound 5 were obtained which were suitable for an X-ray structure analysis. Its molecular structure is depicted in Fig. 3 along with important bond lengths and angles.
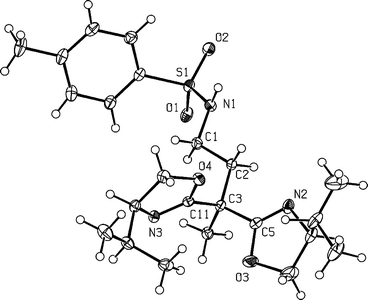 | ||
| Fig. 3 Molecular structure of compound 5. Selected bond lengths (Å) and angles (°): C5–N2, 1.263(3); C11–N3, 1.270(2); N1–S1, 1.6107(16); O1–S1, 1.4376(15); O2–S1, 1.4338(15); C5–C3–C11, 106.21(14); N2–C5–O3, 119.10(19); N2–C5–C3, 127.62(18); N3–C11–O4, 118.57(15); N3–C11–C3, 127.36(16); O1–S1–O2, 120.15(8). | ||
The two oxazoline rings in 5 are twisted relative to one another with the two N-atoms, N2 and N3, adopting opposite orientations. This leads to torsion angles N3–C11–C3–C2 120.3(2)° and C2–C3–C5–N2 8.1(3)°. This particular molecular arrangement is thought to be due to the steric demand of the two isopropyl substituents on the oxazoline rings and differs from that present in the copper complex 10 (vide infra).
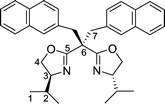
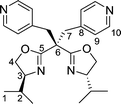
Tetradentate bis(oxazoline) ligands have received less attention than their tridentate counterparts, with very few examples of bisoxazolines incorporating two sidearms on the carbon bridge having been synthesized to date. Their preparation can be easily accomplished by treating the bis(oxazolinyl)methane with an excess of sodium hydride and subsequent treatment with the appropriate electrophiles to give the gem-disubstituted bis(oxazoline) ligands 6–8 in good yields (Scheme 2).
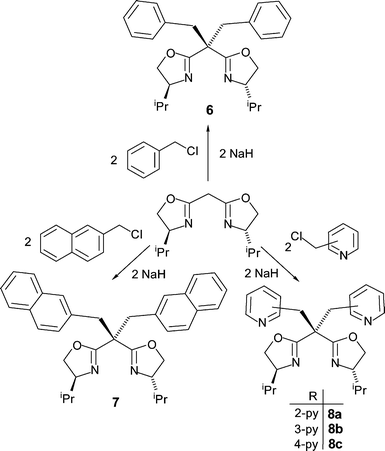 | ||
| Scheme 2 Synthesis of the bisoxazoline ligands 6–8 containing two sidearms. | ||
Asymmetric allylic oxidation of cyclohexene catalyzed by copper complexes containing BOX-derivatives with one and two sidearm(s)
Both the bisoxazoline ligand containing one sidearm as well as those with two were systematically tested as stereodirecting ligands, in combination with a variety of copper salts, in the asymmetric allylic oxidation of cyclohexene which is generally used as the substrate of reference. As the oxidizing agent we employed benzoyl(tert-butyl)peroxide. Previous studies have shown that, using the bisoxazolines BOX or pybox as stereodirecting ligands, enantioselectivities of ca. 60% ee (with yields of ca. 65%) are obtained.Since Andrus and coworkers have reported that it is possible to induce high stereoselectivity (up to 96%, at low conversion) by using the more electron-deficient tert-butyl p-nitroperbenzoate as an oxidant,11 we also tested this reagent. The decreased reactivity of the latter, however, limits the usefulness of the method since only very low yields are obtained. The results obtained with the new ligands 1–8 are summarized in Table 1.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Copper salt | T/°C | Solvent | Yield (%) | ee (%) |
| 1 | 1b | Cu(OTf)2 | 20 | MeCN | 49 | 55 |
| 2 | 1a | Cu(OTf)2 | 20 | MeCN | 52 | 60 |
| 3 | 1a | Cu(BF4)2 | 20 | MeCN | 47 | 54 |
| 4 | 1d | Cu(OTf)2 | 20 | MeCN | 53 | 60 |
| 5 | 1c | Cu(OTf)2 | 20 | MeCN | 41 | 50 |
| 6 | 2a | Cu(OTf)2 | 20 | MeCN | 44 | 51 |
| 7 | 1e | Cu(OTf)2 | 20 | MeCN | 55 | 60 |
| 8 | 2b | Cu(OTf)2 | 20 | MeCN | 45 | 51 |
| 9 | 3a | Cu(OTf)2 | 20 | MeCN | 44 | 61 |
| 10 | 3b | Cu(OTf)2 | 20 | MeCN | 54 | 68 |
| 11 | 4 | Cu(OCl4)2 | 20 | MeCN | 45 | 65 |
| 12 | 4 | Cu(BF4)2 | 20 | MeCN | 72 | 60 |
| 13 | 5 | Cu(OTf)2 | 20 | MeCN | 70 | 60 |
| 14 | 5 | Cu(OCl4)2 | 20 | MeCN | 72 | 60 |
| 15 | 5 | Cu(BF4)2 | 20 | MeCN | 72 | 60 |
| 16 | 5 | Cu(OTf)2 | 20 | Acetone | 59 | 68 |
| 17 | 5 | Cu(OCl4)2 | 20 | Acetone | 67 | 70 |
| 18 | 5 | Cu(BF4)2 | 20 | Acetone | 54 | 70 |
| 19 | 5 | Cu(OTf)2 | 0 | MeCN | 15 | 71 |
| 20 | 5 | Cu(OCl4)2 | 0 | MeCN | 20 | 71 |
| 21 | 5 | Cu(BF4)2 | 0 | MeCN | 20 | 71 |
| 22 | 6 | Cu(OTf)2 | 20 | MeCN | 55 | 80 |
| 23 | 7 | Cu(OTf)2 | 20 | MeCN | 52 | 77 |
| 24 | 8a | Cu(OTf)2 | 20 | MeCN | 61 | 79 |
| 25 | 8a | Cu(OTf)2 | 0 | MeCN | 59 | 77 |
| 26 | 8b | Cu(OTf)2 | 20 | MeCN | 54 | 55 |
| 27 | 8c | Cu(OTf)2 | 20 | MeCN | 45 | 72 |
The complexes which were tested catalyze the reaction with a moderate degree of selectivity and reactivity at relatively low catalyst loading (5 mol%). The type of copper salt employed does not significantly affect the activity and selectivity, whilst the variation of solvent appears to have an effect (with acetone leading to higher selectivities but lower yields than the commonly used acetonitrile). This observation may be attributed to the solvent dependence of cation–anion interaction in the catalyst.
It is worth noting that the copper complexes of ligands 6 and 8a, both bearing two substituents on the carbon atom bridging the two oxazoline units (6: benzyl; 8a: 2-pyridyl), display higher selectivity. As previously observed, the use of p-NO2PhCO3-t-Bu significantly enhances the selectivity of the conversion, albeit with decreased reaction rate (Table 2).11 Compared to previous catalysts tested for the Karasch–Sosnovski reaction, the ee values obtained with the catalysts bearing ligands 6 and 8a are remarkably high (up to 85%). Both ligands differ from one another in functionality and thus potential denticity, however, they are essentially similar as far as the sterics of the bridgehead substituent are concerned.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Copper salt | T/°C | Yield (%) | ee (%) |
| 1 | 5 | Cu(OTf)2 | 20 | 87 | 77 |
| 2 | 5 | Cu(OCl4)2 | 20 | 63 | 77 |
| 3 | 5 | Cu(BF4)2 | 20 | 73 | 72 |
| 4 | 5 | Cu(OTf)2 | −15 | 19 | 79 |
| 5 | 6 | Cu(OTf)2 | 20 | 52 | 85 |
| 6 | 7 | Cu(OTf)2 | 20 | 42 | 84 |
| 7 | 8a | Cu(OTf)2 | 20 | 60 | 82 |
| 8 | 8a | Cu(OTf)2 | 0 | 47 | 85 |
| 9 | 8b | Cu(OTf)2 | 20 | 49 | 65 |
This observation and that of the lower selectivity observed for the catalysts bearing bisoxazoline ligands with only only sidearm function may suggest that the steric demand of the sidearms rather than binding interaction with the metal or the substrates is responsible for the selectivity of the reaction.
Molecular structures of copper(II) complexes containing BOX-derivatives with one and two sidearms
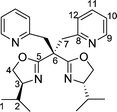
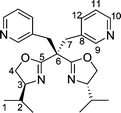
Reaction of copper(II) acetate monohydrate (2 molar equiv.) with the ligands 3a, 5 and 8a (1 molar equiv.) gave the complexes [(ligand)Cu(acetate)2] 9, 10 and 11, respectively (Scheme 3). Single crystals were obtained from toluene solutions layered with n-hexane which were subjected to slow diffusion.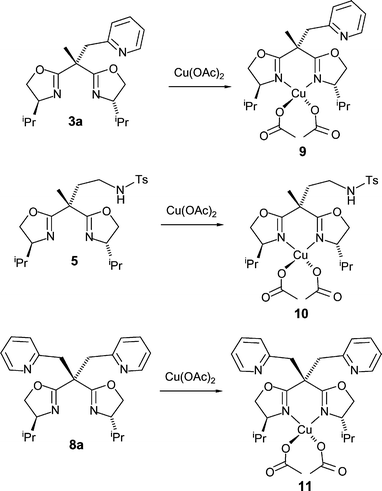 | ||
| Scheme 3 Synthesis of the bisoxazoline–copper(II) complexes 9–11. | ||
The coordination geometry around the copper atoms is approximately octahedral in all three cases (Figs. 4–6). In the elongated distorted CuN2O4 octahedra, the two nitrogen atoms of the oxazolines and one oxygen atom of each acetate occupy the ‘equatorial’ positions. Here, Cu–N and Cu–O distances range from 1.947(6)–1.991(4) Å, respectively. The remaining two oxygen atoms of the acetate ligands approach the ‘axial’ positions, with much larger distances to copper (2.440(3)–2.750(4) Å), and O–Cu–O angles considerably less than 180° (140.8(2)–155.8(1)°). This asymmetrical κ2-acetate coordination is quite common in copper acetate complexes.12 Most significantly, the additional nitrogen donors (weakly nucleophilic sulfonamide NH in 10, more strongly nucleophilic pyridine nitrogen atoms in 9 and 11) do not coordinate to the respective copper centres, nor does there seem to be any significant interaction with the acetato units.
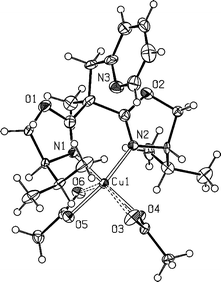 | ||
| Fig. 4 Molecular structure of compound 9. Selected bond lengths (Å) and angles (°): Cu1–N1, 1.946(6); Cu1–N2, 1.973(6); Cu1–O4, 1.954(5); Cu1–O5, 1.977(5); Cu1–O3, 2.687(6); Cu1–O6, 2.540(5); N1–Cu1–N2, 90.1(3); O4–Cu1–N2, 93.9(3); N1–Cu1–O5, 92.4(3); N1–Cu1–O4, 158.2(2); N2–Cu1–O5, 154.1(2); O4–Cu1–O5, 93.2(2). | ||
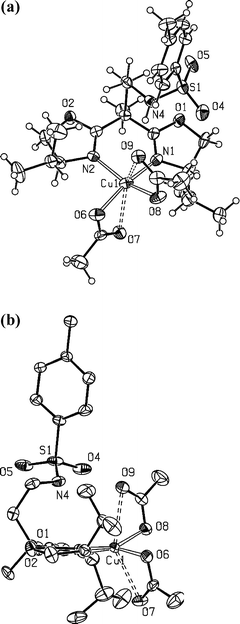 | ||
| Fig. 5 a) Molecular structure of compound 10. Selected bond lengths (Å) and angles (°): Cu1–N1, 1.974(4); Cu1–N2, 1.975(4); Cu1–O6, 1.962(4); Cu1–O8, 1.966(4); N4–S1, 1.603(4); O4–S1, 1.434(4); O5–S1, 1.431(4); Cu1–O7, 2.501(3); Cu1–O9, 2.584(3); N1–Cu1–N2, 89.27(14); O8–Cu1–N1, 91.93(16); O6–Cu1–N2, 92.77(16); O8–Cu1–N2, 157.79(14); O6-Cu1–N1, 162.32(14); O6–Cu1–O8, 92.77(15). b) In-plane view of the complex illustrating the orientation of the tosylamido sidearm (hydrogen atoms omitted for clarity). | ||
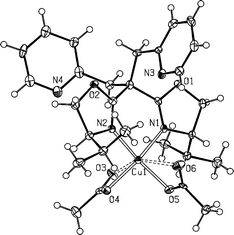 | ||
| Fig. 6 Molecular structure of compound 11. Selected bond lengths (Å) and angles (°): Cu1–N1, 1.991(4); Cu1–N2, 1.976(4); Cu1–O4, 1.986(3); Cu1–O5, 1.948(3); N2–Cu1–N1, 89.72(15); N2–Cu1–O4, 92.80(15); O5–Cu1–N1, 95.75(14); O4–Cu1–N1, 156.45(14); O5–Cu1–N2, 153.24(16); O5–Cu1–O4, 92.48(14). | ||
Conclusions
We have studied the influence of “sidearms” in bisoxazoline copper complexes on their catalyst performance in the asymmetric allylic alkylation of cyclohexene as substrate of reference. In general, the observed enantioselectivities have been somewhat higher than for the unfunctionalized bisoxazoline (“BOX”) derivatives. Notably, this effect was observed regardless of the functional groups incorporated into the sidearm unit, the ee's obtained for the different derivatives being essentially indistinguishable. This leads us to propose that the sidearms do not interfere directly in this reaction, but most probably only play an indirect role by virtue of their steric demand. In other words, we believe that in the case at hand, we observe the third case (c) depicted in Fig. 1. This view is supported by the absence of any interaction of the sidearms with the metal centres or the other ligands established in the X-ray structure analyses of compounds 9–11. Whether this situation pertains for complexes with other transition metals is not clear and under current investigation.Experimental
General
All manipulations were performed under nitrogen. Solvents were dried according to standard methods and saturated with nitrogen. The deuterated solvents used for the NMR spectroscopic measurements were degassed by three successive “freeze–pump–thaw” cycles and stored over 4-Å molecular sieves. All other chemicals were used as received. Yields are given for isolated products showing one spot on a TLC plate and no impurities were detectable in the NMR spectrum. 2,2-Bis((S)-4-isopropyl-4,5-dihydrooxazol-2-yl)-ethane,13 2,2-bis((S)-4-phenyl-4,5-dihydrooxazol-2-yl)-ethane,13 bis((S)-4-isopropyl-4,5-dihydrooxazol-2-yl)-methane,10 2-(2,2-bis((S)-4-isopropyl-4,5-dihydrooxazol-2-yl)propyl)pyridine5 (3a), (4S)-2,2′-(1-phenylmethyl-2-phenylethylidene)bis[4-(1-methylethyl)-4,5-dihydroxazole14 (6) and tert-butyl p-nitrobenzoate11 were prepared according to literature procedures. The 1H and 13C NMR spectra were recorded on Bruker Avance DRX 200 or Varian Unity Plus 400 FT-NMR spectrometers at room temperature (295 K). The spectra were referenced relative to tetramethylsilane using the residual protio-solvent (1H) or the carbon (13C) resonance. IR spectra were recorded as thin films between KBr plates using a FT-IR Merlin Excalibur FT3000 instrument. Mass spectra (EI or FAB) were recorded on Finnigan MAT TSQ 700 or JEOL JMS-700 spectrometers, high resolution mass spectra (HRMS) were recorded on the latter instrument. Elemental analyses were performed by the microanalytical service at the chemistry department of Heidelberg. Enantiomeric excesses were determined by using a Finnigan Surveyer™ Modular HPLC System equipped with a Chiralpak® AS–H and/or Chiralcel® AD–H columns.Preparation of the compounds
General procedure for the asymmetric allylic oxidation catalyzed by Cu(II) complexes
A solution of the suitable ligand (0.05 mmol) and Cu salt (0.04 mmol) in distilled acetonitrile (3.0 mL) was stirred a rt for 1 h to ensure the formation of the copper complex. The solution was then cooled to the desired temperature and the cyclohexene (5 mmol) was added. To the stirred mixture a solution of peroxyester (0.85 mmol) in acetonitrile (2 mL) was added dropwise. After the reaction was judged to be complete (TLC disappearance of the peroxyester), which generally was the case after 5–7 days, the solvent was removed in vacuo, and the residue was dissolved in CH2Cl2 (20 mL), washed successively with an aqueous KHCO3 solution (10%), brine, and water and dried over Na2SO4. Concentration and chromatography on a silica gel column with a hexane–ethyl acetate mixture (50 : 1) afforded pure allylic benzoate. The yields and ee's are given in Tables 1 and 2. In general, slightly higher yields were obtained upon increasing the reaction times without loss of enantioselectivityThe enantiopurity of the products was determined by chiral HPLC using the following conditions:
Crystal structure determinations
Suitable crystals of the protioligand 5 and the complexes 9·H2O, 10 and 11 were obtained by layering concentrated solutions in dichloromethane with pentane or diethyl ether and allowing slow diffusion at room temperature. Intensity data were collected at low temperature on a Bruker AXS Smart 1000 CCD (5, 9, 10) and a Nonius Kappa CCD (11) diffractometer. A semi-empirical absorption correction was applied. The structures were solved with heavy atom and/or direct methods and refined by full matrix least squares. Hydrogen atoms were input at calculated positions and refined with the riding model, except the amide hydrogens, which were taken from difference Fourier synthesis and refined. In complex 10, the distance d(N4–H4) was restrained to 0.93(2) Å. The positions of the hydrogen atoms of the water of crystallization in 9·H2O were inferred by relatively short intermolecular O⋯O vectors. The calculations were performed using the programs DIRDIF,15 SHELXS-8616 and SHELXL-97.17 Graphical representations were drawn with PLATON.18 Anisotropic displacement elipsoids are scaled to 40% probability. Crystal and experimental data are given in Table 3.CCDC reference numbers 283311–283314.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512570g
| 5 | 9·H2O | 10 | 11 | |
|---|---|---|---|---|
| Empirical formula | C23H35N3O4S | C24H37CuN3O7 | C27H41CuN3O8S | C29H38CuN4O6 |
| Formula weight | 449.60 | 543.11 | 631.22 | 602.17 |
| Crystal size/mm | 0.30 × 0.25 × 0.13 | 0.22 × 0.06 × 0.02 | 0.25 × 0.25 × 0.10 | 0.30 × 0.25 × 0.15 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | P21 | P212121 | P21 | P212121 |
| a/Å | 6.5200(4) | 9.3854(12) | 9.3203(6) | 9.1940(10) |
| b/Å | 19.3181(11) | 14.6399(18) | 10.7524(8) | 16.122(2) |
| c/Å | 9.6743(6) | 19.322(2) | 16.0394(11) | 19.152(6) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 106.0890(10 | 90 | 104.198(2 | 90 |
| γ/° | 90 | 90 | 90 | 90 |
| V/Å3 | 1170.79(12) | 2654.9(6) | 1558.30(19) | 2838.8(10) |
| Z | 2 | 4 | 2 | 4 |
| D c/Mg m−3 | 1.275 | 1.359 | 1.345 | 1.409 |
| μ/mm−1 | 0.172 | 0.869 | 0.817 | 0.819 |
| T/K | 100(2) | 100(2) | 100(2) | 193(2) |
| F(000) | 484 | 1148 | 666 | 1268 |
| Refl. collected | 18426 | 29150 | 13654 | 10824 |
| Refl. indep. [Rint] | 6926 [0.035] | 4693 [0.137] | 6927 [0.053] | 4819 [0.061] |
| Data/rest./par. | 6926/1/302 | 4693/0/323 | 6927/1/367 | 4819/0/367 |
| Goodness-of-fit on F2 | 1.042 | 1.054 | 1.057 | 1.095 |
| Final R indices [I > 2σ(I)] | R 1 = 0.045, wR2 = 0.106 | R 1 = 0.074, wR2 = 0.191 | R 1 = 0.053, wR2 = 0.129 | R = 0.047, wR2 = 0.089 |
| R indices (all data) | R 1 = 0.060, wR2 = 0.116 | R 1 = 0.092, wR2 = 0.203 | R 1 = 0.072, wR2 = 0.138 | R = 0.069, wR2 = 0.108 |
| Largest residual peak and hole/e Å−3 | 0.69 and −0.36 | 0.93 and −0.74 | 1.47 and −0.88 | 0.38 and −0.48 |
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft for support of this work, the Alexander-von-Humboldt Foundation for a fellowship (C.C.) and Matthieu Eckert for experimental help. Support by BASF (Ludwigshafen) and Degussa (Hanau) is gratefully acknowledged.References
- (a) R. E. Lowenthal, A. Abiko and S. Masamune, Tetrahedron Lett., 1990, 31, 6005 CrossRef CAS; (b) D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726 CrossRef CAS; (c) E. J. Corey, N. Imai and H. Y. Zhang, J. Am. Chem. Soc., 1991, 113, 728 CrossRef CAS; (d) D. Müller, G. Umbricht, B. Weber and A. Pfaltz, Helv. Chim. Acta, 1991, 74, 232 CrossRef.
- Selected examples: (a) D. A. Evans, D. Seidel, M. Rueping, H. W. Lam, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2003, 125, 12692 CrossRef CAS; (b) D. A. Evans, K. A. Scheidt, J. N. Johnston and M. C. Willis, J. Am. Chem. Soc., 2001, 123, 4480 CrossRef CAS; (c) J. Cong-Dung Le and B. Pagenkopf, Org. Lett., 2004, 6, 4097 CrossRef CAS; (d) J. Thorhauge, M. Johannsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 1998, 37, 2404 CrossRef CAS; (e) D. A. Evans and J. M. Janey, Org. Lett., 2001, 3, 2125 CrossRef CAS; (f) C. Christensen, K. Juhl and K. A. Jørgensen, Chem. Commun., 2001, 2222 RSC; (g) D. A. Evans, J. S. Johnson and E. J. Olhava, J. Am. Chem. Soc., 2000, 122, 1635 CrossRef CAS; (h) F. Glorius and A. Pfaltz, Org. Lett., 1999, 1, 141 CrossRef CAS.
- Reviews: (a) F. Fache, E. Schulz, M. Lorraine Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159 CrossRef CAS; (b) A. K. Ghosh, P. Mathivanan and J. Cappiello, Tetrahedron: Asymmetry, 1998, 9, 1 CrossRef CAS; (c) H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151 CrossRef CAS.
- (a) J. Zhou and Y. Tang, J. Am. Chem. Soc., 2002, 124, 9030 CrossRef CAS; (b) J. Zhou, M.-C. Ye, Z.-Z. Huang and Y. Tang, J. Org. Chem., 2004, 69, 1309 CrossRef CAS; (c) J. Zhou and Y. Tang, Chem. Commun., 2004, 432 RSC; (d) M.-C. Ye, B. Li, J. Zhou, X.-L. Sun and Y. Tang, J. Org. Chem., 2005, 70, ASAP.
- Y. Tang, M. C. Ye and J. Zhou, J. Comb. Chem., 2004, 6, 301 CrossRef CAS.
- (a) S. Bellemin-Laponnaz and L. H. Gade, Chem. Commun., 2002, 1286 RSC; (b) C. Dro, S. Bellemin-Laponnaz, R. Welter and L. H. Gade, Angew. Chem., Int. Ed., 2004, 43, 4479 CrossRef CAS; (c) B. D. Ward, S. Bellemin-Laponnaz and L. H. Gade, Angew. Chem., Int. Ed., 2005, 44, 1668 CrossRef CAS; (d) C. Foltz, B. Stecker, G. Marconi, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Chem. Commun., 2005, 5115 RSC.
- M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1958, 80, 756 CrossRef CAS.
- (a) M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1959, 81, 5819 CrossRef CAS; (b) A. L. Beckwith and A. A. Zavistas, J. Am. Chem. Soc., 1986, 108, 8230 CrossRef CAS.
- (a) A. S. Gokhale, A. B. E. Minindis and A. Pfaltz, Tetrahedron Lett., 1995, 36, 1831 CrossRef CAS; (b) M. B. Andrus, A. B. Agrade, X. Chen and M. G. Pamment, Tetrahedron Lett., 1995, 36, 2945 CrossRef CAS; (c) K. Kawasaki, S. Tsumura and T. Katsuki, Synlett, 1995, 1245 CrossRef CAS; (d) A. Levina and J. Muzart, Tetrahedron: Asymmetry, 1995, 6, 147 CrossRef CAS. For reviews, see: (e) M. B. Andrus and J. C. Lashley, Tetrahedron, 2002, 58, 845 CrossRef CAS; (f) J. Eames and M. Watkinson, Angew. Chem., Int. Ed., 2001, 40, 3567 CrossRef CAS.
- S. E. Denmark and C. M. Stiff, J. Org. Chem., 2000, 98, 5875 CrossRef.
- M. B. Andrus and Z. Zhou, J. Am. Chem. Soc., 2002, 124, 8806 CrossRef CAS.
- (a) J. Ratilainen, K. Airola, R. Fröhlich, M. Nieger and K. Rissanen, Polyhedron, 1999, 18, 2265 CrossRef CAS; (b) A. S. Antsyshkina, M. A. Porai-Koshits, V. N. Ostrikova, M. Pilkington and H. Stoeckli-Evans, Eur. J. Inorg. Chem., 2002, 1985; (c) C. P. Raptopoulou, S. Paschalidou, A. A. Pantazaki, A. Terzis, S. P. Perlepes, T. Lialiaris, E. G. Bakalbassis, J. Mrozinski and D. A. Kyriakidis, J. Inorg. Biochem., 1998, 71, 15 CrossRef CAS; (d) P. Molenveld, J. F. J. Engbersen, H. Kooijman, A. L. Spek and D. N. Reinhoudt, J. Am. Chem. Soc., 1998, 120, 6726 CrossRef CAS; (e) S. Youngme, C. Pakawatchai, H.-K. Fun and K. Chinnakali, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1586 CrossRef; (f) B. Kozlevcar, A. Murn, K. Podlipnik, N. Lah, I. Leban and P. Segedin, Croat. Chem. Acta, 2004, 77, 613 CAS; (g) R. Grobelny, T. Glowiak, J. Mrozinski, L. Brzozka, W. Baran and P. Tomasik, Pol. J. Chem., 1995, 69, 559 CAS; (h) M. B. Meder and L. H. Gade, Eur. J. Inorg. Chem., 2004, 2716 CrossRef CAS.
- S. Bellemin-Laponnaz and L. H. Gade, Angew. Chem., Int. Ed., 2002, 41, 3473 CrossRef CAS.
- M. Honma, T. Sawada, Y. Fujisawa, M. Utsugi, H. Watanabe, A. Umino, T. Matsamura, T. Hagihara, M. Takano and M. Nakada, J. Am. Chem. Soc., 2003, 125, 2860 CrossRef CAS.
- P. T. Beurskens, G. Beurskens, R. de Gelder, S. G. Granda, R. O. Gould, R. Israel and J. M. M. Smits, DIRDIF-99, University of Nijmegen, The Netherlands, 1999 Search PubMed.
- G. M. Sheldrick, SHELXS-86, University of Göttingen, Germany, 1986 Search PubMed.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |