Ligand dependence of π-complex character in disilene–palladium complexes†
Takeaki
Iwamoto
,
Yumiko
Sekiguchi
,
Naoki
Yoshida
,
Chizuko
Kabuto
and
Mitsuo
Kira
*
Department of Chemistry, Graduate School of Science, Tohoku University, Aoba-ku, Sendai, Japan. E-mail: mkira@si.chem.tohoku.ac.jp; Fax: +81-22-795-6589; Tel: +81-22-795-6585
First published on 15th November 2005
Abstract
The synthesis and structures of new 16-electron disilene palladium complexes with 2,6-dimethylphenyl isocyanide and phenyldimethylphosphine ligands [L1L2Pd{(t-BuMe2Si)2Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(SiMe2Bu-t)2}, where L1 = L2 = PhMe2P; L1 = (cyclohexyl)3P, L2 = 2,6-dimethylphenyl isocyanide; L1 = L2 = 2,6-dimethylphenyl isocyanide] are described. Comparison of the X-ray structural parameters around the disilene moiety among these complexes and related bis(trimethylphosphine)(disilene)palladium and 14-electron (tricyclohexylphosphine)(disilene)palladium revealed that the π-complex character is sensitive to the residual ligands and increases with decreasing the strength of σ-donation from the ligands.
Si(SiMe2Bu-t)2}, where L1 = L2 = PhMe2P; L1 = (cyclohexyl)3P, L2 = 2,6-dimethylphenyl isocyanide; L1 = L2 = 2,6-dimethylphenyl isocyanide] are described. Comparison of the X-ray structural parameters around the disilene moiety among these complexes and related bis(trimethylphosphine)(disilene)palladium and 14-electron (tricyclohexylphosphine)(disilene)palladium revealed that the π-complex character is sensitive to the residual ligands and increases with decreasing the strength of σ-donation from the ligands.
Introduction
The bonding of an alkene to a transition metal center in alkene transition metal complexes1 is usually understood in terms of alkene-to-metal σ-donation and metal-to-alkene π-back donation, according to the Dewar–Chatt–Duncanson model.2 The structure of an alkene complex is significantly influenced by the relative importance between the σ-donation and the π-back donation, and hence, they are classified into either a π-complex having major contribution of σ-donation or a metallacycle having dominant π-back donation. The geometry around the alkene ligand in the π-complex is not very much different from that of the free alkene, while that in the metallacycle is significantly distorted and characterized by an elongated C1–C2 bond length (d) and a large bent angle (θ) defined by the angle between the R1–C1–R2 (or R3–C2–R4) plane and the C1–C2 bond (Chart 1).3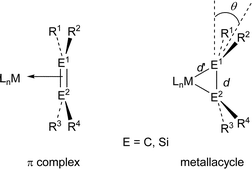
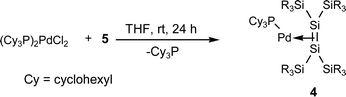
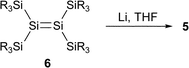
Much attention has been focused recently on the structure of transition metal complexes with η2-silicon–silicon doubly bonded species as ligands (disilene complexes).4–8 A variety of isolable η2-disilene complexes of platinum,4,5a palladium,5b,c tungsten,6 molybdenum,6 iron,7 and zirconium8 have been synthesized and some of them have been investigated by X-ray crystallography. We have recently synthesized 16-electron platinum and palladium complexes with a silyl-substituted η2-disilene ligand 1–35a,b and 14-electron disilene complex 45c using the reactions of the corresponding bis(phosphine)dichlorometals with 1,2-dilithiotetrakis(tert-butyldimethylsilyl)disilane 5,9 which was prepared by the reaction of stable disilene 610 with lithium in THF (eqn (1)–(3)).
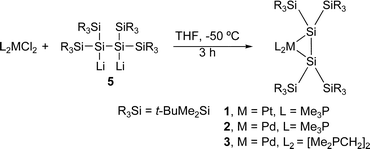 | (1) |
 | (2) |
 | (3) |
In contrast to alkene complexes, most of the disilene complexes whose X-ray structures are known4–7 are characterized as metallacycles, by applying the Dewar–Chatt–Duncunson type bonding scheme to the disilene complexes. However, the character may depend on the central metal, ligands, and substituents on disilene. Actually, we have found that 16-electron disilene palladium complex 2 has slightly larger π-complex character than the corresponding platinum complex 1.5b The first 14-electron disilene palladium complex 4 has stronger π-complex character than the related 16-electron complexes 2 and 3.5c
We wish herein to report the synthesis and structures of new 16-electron disilene palladium complexes with 2,6-dimethylphenyl isocyanide (7) and phenyldimethylphosphine as the residual ligands. Comparison of the structural parameters around the disilene moiety revealed that the π-complex character depends remarkably on the residual ligands.
Results and discussion
Synthesis of new 16-electron disilene palladium complexes
Very few reactions of disilene transition metal complexes have been reported to date. Disilene–platinum complexes synthesized by Pham and West react with small molecules like oxygen, hydrogen, and methanol to afford the corresponding Si–Si or Si–Pt bond cleavage products.4 Berry et al. have revealed that the Si–Si and Si–metal bonds of molybdenum and tungsten complexes of tetramethyldisilene show high reactivity toward methanol, carbon dioxide and triphenylphosphine sulfide.6 However, there have been no ligand exchange reactions of disilene complexes reported. We have found the reactions of disilene palladium complexes 2 and 4 with isocyanide 7 to afford the corresponding disilene complexes with isocyanide ligands; the disilene ligand remains intact during the reactions.The reactions of 14-electron complex 4 with 1 and 2 equivalents of isocyanide 7 gave mono(isocyanide) complex 8 and bis(isocyanide) complex 9, respectively (Scheme 1).
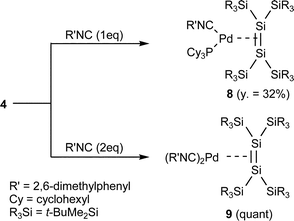 | ||
| Scheme 1 Synthesis of disilene complexes with isocyanide ligands. | ||
Interestingly, the reactions of 16-electron complex 2 with 1 and 2 equivalents of isocyanide 7 also gave new mono(isocyanide) complex 11 in 83% isolation yield and 9 in quantitative yield, respectively. The result suggests that 16-electron complex 2 exists in equilibrium with 14-electron complex 10 and dissociated trimethylphosphine (eqn. (4) and (5)); the subsequent dissociation of trimethylphosphine from 11 followed by the addition of the isocyanide will give bis(isocyanide) complex 9. Because no 14-electron complexes 10 and 12 were detected in solution by NMR spectroscopy, the concentration of 10 and 12 in the equilibrium will be very low.
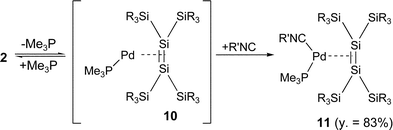 | (4) |
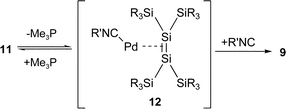 | (5) |
To elucidate the electronic effects of phosphine ligands on the π-complex character of the 16-electron complex 2, complex 13 was synthesized by the conventional method5,11 (eqn (6)).
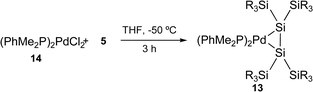 | (6) |
Molecular structures of 16-electron disilene complexes
Molecular structures of disilene palladium complexes 8, 9 and 13 determined by X-ray crystallography are shown in Fig. 1–3. The structural parameters are listed in Table 1 together with those for disilene complexes 2, 3 and 4.12 Because mono(isocyanide) complex 8 showed two crystallographically independent molecules in an asymmetric unit, two sets of parameters are shown in the table.|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | L1 | L2 | d/Å | Δd/Å (%Δd/d0)a | θ 1/° | θ 2/° | θ av b/° | d 1/Å | d 2/Å | φ c/° | δ(29Si) |
|
a
d
0 is the SiSi distance in free (t-BuMe2Si)2SiSi(SiMe2Bu-t)2 (2.202 (1) Å).10
Δd = d
−
d0 = d
− 2.202. Standard deviations of %Δd/d0 and θ are estimated to be at most 0.13 and 0.05°, respectively.
b
θ
av = (θ1 + θ2)/2.
c Dihedral angle between plane (L1–Pd–L2) and plane (Si–Pd–Si).
d R′ = 2,6-dimethylphenyl.
|
|||||||||||
| 2 | PMe3 | PMe3 | 2.3027(8) | 0.101 (5.2) | 27.2 | 27.2 | 27.2 | 2.4369(5) | 2.4369(5) | 31.8 | −46.5 |
| 3 | Me2PCH2CH2PMe2 | 2.3180(8) | 0.118 (4.6) | 27.3 | 27.3 | 27.3 | 2.4184(4) | 2.4184(4) | 28.5 | −51.9 | |
| 13 | PhMe2P | PhMe2P | 2.2952(13) | 0.093 (4.2) | 14.4 | 26.8 | 20.6 | 2.4632(10) | 2.4287(10) | 31.0 | −44.8 |
| 8 (mol. 1) | R′NCd | Cy3P | 2.2861(11) | 0.085 (3.9) | 5.2 | 20.6 | 12.9 | 2.4597(8) | 2.4581(9) | 36.4 | −39.4, −60.4 |
| 8 (mol. 2) | R′NCd | Cy3P | 2.2967(11) | 0.094 (4.3) | 3.0 | 36.5 | 19.8 | 2.4603(8) | 2.4264(8) | 31.1 | — |
| 9 | R′NCd | R′NCd | 2.289(2) | 0.087 (4.0) | 9.5 | 8.9 | 9.2 | 2.4340(11) | 2.4411(11) | 20.0 | −41.2 |
| 4 | Cy3P | — | 2.273(1) | 0.072 (3.3) | 4.4 | 9.7 | 7.0 | 2.3610(9) | 2.4168(8) | — | +65.3 |

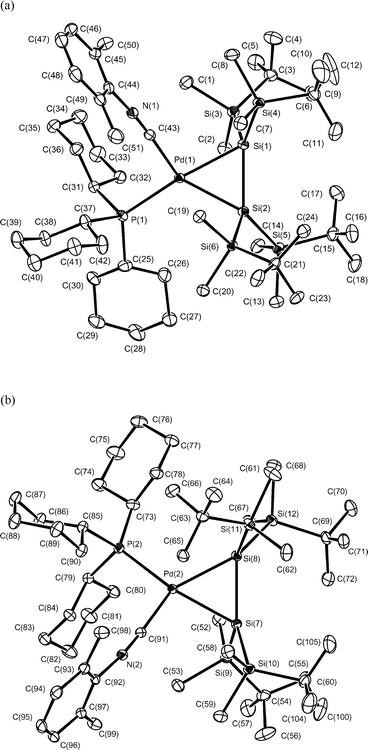 | ||
| Fig. 1 Molecular structure of complex 8. Two crystallographically independent molecules were observed in an asymmetric unit: (a) molecule 1, (b) molecule 2. Thermal ellipsoids are shown in the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Pd(1)–Si(1) 2.4597(8), Pd(1)–Si(2) 2.4581(9), Si(1)–Si(2) 2.2861(11), Pd(1)–P(1) 2.4437(8), Pd(1)–C(43) 1.989(3); Si(1)–Pd(1)–Si(2) 55.40(3), Pd(1)–Si(1)–Si(2) 62.26(3), Pd(1)–Si(2)–Si(1) 62.33(3), P(1)–Pd(1)–C(43) 96.06(9), Pd(2)–Si(7) 2.4603(8), Pd(2)–Si(8) 2.4264(8), Si(7)–Si(8) 2.2967(11), Pd(2)–P(2) 2.4352(8), Pd(2)–C(91) 2.001(3), Si(7)–Pd(2)–Si(8) 56.06(3), Pd(2)–Si(7)–Si(8) 61.22(3), Pd(2)–Si(8)–Si(7) 62.72(3), P(2)–Pd(2)–C(91) 96.48(9). | ||
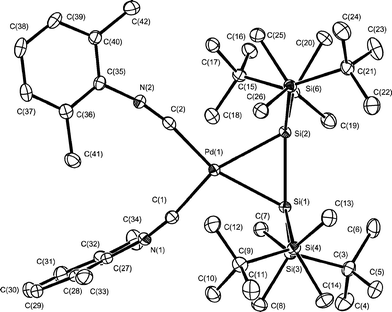 | ||
| Fig. 2 Molecular structure of complex 9. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Pd(1)–Si(1) 2.4340(11), Pd(1)–Si(2) 2.4411(11), Si(1)–Si(2) 2.2891(14), Pd(1)–C(1) 2.014(4), Pd(1)–C(2) 2.029(4); Si(1)–Pd(1)–Si(2) 56.01(4), Pd(1)–Si(1)–Si(2) 62.15(4), Pd(1)–Si(2)–Si(1) 61.84(4), C(1)–Pd(1)–C(2) 97.59(15). | ||
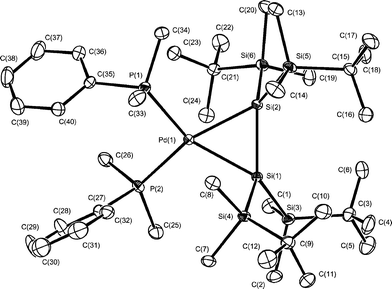 | ||
| Fig. 3 Molecular structure of complex 13. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Pd(1)–Si(1) 2.6432(10), Pd(1)–Si(2) 2.4287(10), Si(1)–Si(2) 2.2952(13), Pd(1)–P(1) 2.3574(10), Pd(1)–P(2) 2.3566(10); Si(1)–Pd(1)–Si(2) 55.96(3), Pd(1)–Si(1)–Si(2) 61.26(3), Pd(1)–Si(2)–Si(1) 62.78(3), P(1)–Pd(1)–P(2) 101.14(4). | ||
The Si(1)–Si(2) bond distances (d) of all the disilene palladium complexes (2.273–2.3180 Å) lie below the shortest limit of the reported Si–Si single bond (2.335–2.697 Å) but depend significantly on the ligands.13 The central palladium atom of the disilene complexes 8, 9 and 13 adopts highly distorted square planar geometry with the angle between Si1–Pd–Si2 and L1–Pd–L2 planes (φ in Table 1) of 36.4 (31.1), 20.0 and 31.0°, respectively; the angle φ for 2 and 3 is 31.8 and 28.5°, respectively,5b while the geometry around palladium in the 14-electron disilene complex 4 is almost planar.5d Steric repulsion between bulky trialkylsilyl substituents and other ligands could be responsible for the distorted geometry. The extent of the steric repulsion for 9 with less bulky isocyanide ligands is much smaller than that for other 16-electron disilene complexes. The Si–Pd distances (d1 and d2, 2.360–2.460 Å) are a little longer than those of reported bis(phosphine)(disilyl)palladium complexes (2.34–2.38 Å).14
The bent angles (θ1 and θ2), which are defined as an angle between the Si1(2)–Si2(1)–R plane and the Si1–Si2 bond, are larger than that of free disilene 6 (0.1°)10 but the extent depends remarkably on the ligands. The bond elongation estimated by Δd/d0 values (Δd = d − d0, where d0 is the Si1–Si2 bond length of free disilene 610), is not very sensitive to the ligands on palladium but decreases in the order of 3 > 2 > 13 > 8 (av.) ∼ 9 > 4. On the other hand, the averaged bent angles (θav.) are significantly affected by the ligands and decrease in almost the same order as the bond elongation; 3 ∼ 2 > 13 > 8 (av.) > 9 > 4.15
If the Dewar–Chatt–Duncanson model is applicable to disilene complexes, the Δd/d0 value and θ value should decrease with increasing the π-complex character of the disilene complexes. On the basis of this criterion, the π-complex character is evaluated to decrease in the following order: 4 > 9 > 8 (av.) > 13 > 2 ∼ 3. As shown in a previous paper,5c the π-back donation in 14-electron disilene complex 4 is the smallest because one basic ligand on palladium is missing. The σ-donor ability of ligands is expected to decrease in the order of Me3P > PhMe2P > R′NC on the basis of the basicity of the ligands. Thus, the π-back donation from the metal will increase in the order of 4 < 9 < 8 < 13 < 2 ∼ 3. As expected, the order is in good accord with the observed order for the π-complex character.
The π-complex character of unsymmetrically substituted 16-electron disilene complex 8 is between those of 2 and 9, suggesting the additivity of the extent of the σ-donation to the metal. It is expected that because of stronger trans influence of phosphine than isocyanide, the bent angle around an unsaturated silicon atom of complex 8 located at the trans position (θ1) to tricyclohexylphosphine should be much larger than that for the other unsaturated silicon atom (θ2). In reality, θ1 and θ2 in complex 8 are 5.2 (3.0) and 20.6 (36.5)°, respectively; θ1 is much smaller than θ2, in contrast to the above expectation. The incompatibility between the expectation and the experimental results should not be ascribed to the steric effects of bulky tricyclohexylphosphine, because the geometrical characteristics for 8 was reproduced qualitatively by the theoretical calculations for a model disilene palladium complexes (vide infra).
Interestingly, even symmetrically substituted disilene complexes 9 and 13 showed two different bent angles and Pd–Si distances. The results may be attributed to the fact that the steric and electronic environments around two unsaturated silicon atoms in these complexes are different to each other in the solid state because the two residual ligands in complex 9 or 13 adopt the different rotational conformations.
To evaluate the steric effects on the bent angles θ1 and θ2, theoretical calculations were performed for model 16-electron disilene–palladium complexes 8′ and 9′ at the B3LYP/6-31G* level (Chart 2).16,17 The θ values for 2′5c8′ and 9′ are 27.9, 25.3 (av.) and 22.4, and %Δd/d0 values for 2′, 8′ and 9′ are 4.3, 4.4 and 4.1, respectively. Despite the geometry around palladium of all these model complexes is planar (torsion angles φ are less than 5°), the orders of θ and %Δd/d0 values are well in accord with those for a set of 2, 8 and 9. The bent angle θav and the bond elongation Δd/d0 may serve as good indices for the π-complex character of disilene complexes, even though disilene complexes are distorted significantly from the ideal square planar geometry.
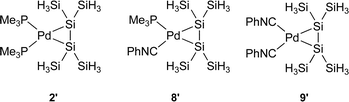 | ||
| Chart 2 | ||
As shown in Table 1, θ1 is unexpectedly much smaller than θ2 for unsymmetrically substituted disilene complex 8. The unsymmetrical distortion is not ascribed to the steric hindrance between ligands, because the tendency was reproduced in less hindered model complex 8′ with θ1 and θ2 of 19.6 and 31.0°, respectively. Because θ1 is larger than θ2 in 14-electron disilene complex 4 whose fourth ligand is missing, sole smaller σ donor ability of aryl isocyanide than trialkylphosphine cannot explain the unsymmetrical distortion in 8 and 8′. Further work is required to elucidate the origin of the unsymmetric bent angles in 8.
We have recently shown 14-electron disilene complexes 4 show the 29Si resonance at very low field of +65.3 ppm compared to those of 16-electron disilene complexes 2 (−46.5 ppm) and 3 (−51.9 ppm) and the very low field resonance was taken to be an indication of the strong π-complex character.5c Although the 29Si resonance of the unsaturated silicon nuclei for 9 (−41.2 ppm) appears at a little lower field than those for 2 and 3, the extent of the down-field shift for 9 is much smaller than that observed for 4. The remarkable low-field shift of the 29Si resonance of 4 is characteristic of 14-electron, three-coordinate complexes.
Conclusion
We have shown that the π-complex character of disilene transition metal complexes is significantly modified by the residual ligands. The π-complex character in L2Pd(disilene)-type 16-electron complexes remarkably increases by changing L from trialkylphosphine to aromatic isocyanide with poorer σ-donating ability, while the π-complex character is smaller than that of the previously reported 14-electron disilene complex having one trialkylphosphine ligand.Experimental
1H, 13C, 29Si and 31P NMR spectra were recorded on a Bruker AC300P FT-NMR spectrometer at 300, 75.4, 59.6 and 121 MHz, respectively, and a Bruker Avance 400P FT-NMR spectrometer at 400, 100, 79 and 161 MHz, respectively. Mass spectra and high resolution mass spectra were recorded on a HX-110 mass spectrometer (FAB, nitrobenzyl alcohol matrix). Air-sensitive compounds were manipulated in a VAC Nexus 100027 glove box. cis-Bis(phenyl isocyanide)dichloropalladium,18 tetrakis(tert-butyldimethylsilyl)disilene (6)10 and 1,2-dilithiotetrakis(tert-butyldimethylsilyl)disilane (5)9 were prepared according to the procedure in the literature.[η2-Tetrakis(tert-butyldimethylsilyl)disilene](2,6-dimethylphenyl isocyanide)(tricyclohexylphosphine)palladium 8
To a mixture of disilene complex 4 (0.070 g, 0.077 mmol) and isocyanide 7 (0.010 g, 0.076 mmol) in a degassed Schlenk flask (50 cm3) was introduced dry THF (5 cm3) through a vacuum line at −50 °C. Increasing the temperature from −50 °C to rt during 4.5 h resulted a clear yellow solution. Evaporation of THF and then recrystallization from toluene at −20 °C afforded mono(isocyanide) complex 8 (0.080 g, 0.025 mmol) in 32% yield. 8: Found: C, 59.07; H, 9.88. C51H102PNPdSi6 requires C, 59.17; H, 9.93%; mp 145 °C (decomp.); δH (400 MHz, C6D6) 0.47 (s, 6 H), 0.51 (s, 6 H), 0.56 (s, 6 H), 0.63 (s, 6 H), 1.23–2.16 (m, 33 H), 1.29 (s, 18 H), 1.33 (s, 18 H), 2.43 (s, 6 H), 6.63 (d, J = 7.6 Hz, 2 H), 6.73 (dd, J = 7.6 Hz, 1 H); δC (100 MHz, THF-d8) 1.1, 1.4, 1.6, 1.7 (SiCH3), 17.7 (CArCH3), 18.5, 18.7 (C(CH3)3), 25.4 (PCHCH2CH2CH2), 26.7 (d, 2JP–C = 10 Hz, PCHCH2), 28.2, 28.3 (C(CH3)3), 30.0 (PCHCH2CH2), 33.0 (d, 1JP–C = 6 Hz, PCH), 126.8 (Cipso), 127.2 (CAr(m)), 127.3 (CAr(p)), 132.6 (CAr(o)), 164.1 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N); δSi (79 MHz, C6D6) −60.4 (d, 2JP(cis)–Si = 15 Hz), −39.4 (d, 2JP(trans)–Si = 48 Hz), +4.97, +4.99, 5.8, 5.9; δP (161 MHz, C6D6) +30.9.
N); δSi (79 MHz, C6D6) −60.4 (d, 2JP(cis)–Si = 15 Hz), −39.4 (d, 2JP(trans)–Si = 48 Hz), +4.97, +4.99, 5.8, 5.9; δP (161 MHz, C6D6) +30.9.
[η2-Tetrakis(tert-butyldimethylsilyl)disilene]bis(2,6-dimethylphenyl isocyanide)palladium 9
To a mixture of disilene complex 4 (7.0 mg, 7.7 × 10−3 mmol) and isocyanide 7 (2.0 mg, 1.5 × 10−2 mmol) in a degassed NMR tube (5 mm ϕ) was introduced dry benzene-d6 (0.4 cm3) through a vacuum line at −50 °C. Quantitative formation of bis(isocyanide) complex 9 was confirmed by 1H, 29Si and 31P NMR spectroscopies. Evaporation of benzene-d6 and recrystallization from toluene at −20 °C gave 9 as orange plates. 9: mp 136 °C (decomp.); δH (400 MHz, C6D6) 0.56 (s, 12 H), 0.59 (s, 12 H), 1.30 (s, 36 H), 2.28 (s, 12 H), 6.65 (d, J = 7.6 Hz, 4 H), 6.77 (dd, J = 7.6 Hz, 2 H); δC (100 MHz, THF-d8) 1.4, 1.6 (SiCH3), 17.5 (CArCH3), 18.1 (C(CH3)3), 28.3 (C(CH3)3), 126.3 (Cipso), 127.1 (CAr(m)), 127.7 (CAr(p)), 133.3 (CAr(o)), 161.6 (CN); δSi (79 MHz, C6D6) −41.2, +6.6.
[η2-Tetrakis(tert-butyldimethylsilyl)disilene](2,6-dimethylphenyl isocyanide)(trimethylphosphine)palladium 11
To a mixture of 16-electron disilene complex 2 (0.100 g, 0.129 mmol) and isocyanide 7 (0.017 g, 0.130 mmol) in a degassed Schlenk flask (50 cm3) was introduced dry toluene (5 cm3) through a vacuum line. Stirring the resulting light orange solution for 16 h at rt, distilling off the solvent, and recrystallization from hexane at −20 °C gave 11 (0.089 g, 0.107 mmol) as orange prisms in 83% yield. 11: δH (400 MHz, C6D6) 0.46 (s, 6 H), 0.47 (s, 6 H), 0.51 (s, 6 H), 0.56 (s, 6 H), 1.23 (d, 2JP–H = 6 Hz, 9 H), 1.25 (s, 18 H), 1.27 (s, 18 H), 2.27 (s, 6 H), 6.67 (d, J = 7.0 Hz, 2 H), 6.76 (dd, J = 7.0 Hz, 1 H); δC (100 MHz, THF-d8) 1.2 (br), 1.3, 1.4 (SiCH3), 17.6 (CArCH3), 18.3 (d, 1JP–C = 3 Hz, PCH3), 18.4, 18.5 (C(CH3)3), 28.2, 28.3 (C(CH3)3), 126.7 (Cipso), 127.2 (CAr(m)), 127.4 (CAr(p)), 132.9 (CAr(o)), 163.1 (CN); δSi (79 MHz, C6D6) −48.0 (d, 2JP(cis)–Si = 17 Hz), −44.7 (d, 2JP(trans)–Si = 60 Hz), 5.8, 5.9, 6.0, 6.1; δP (161 MHz, C6D6) −31.4 (m).
Stepwise ligand exchange of disilene complex 2 with isocyanide 7 followed by NMR spectroscopy
When disilene complex 2 (10 mg, 1.5 × 10−5 mol) and isocyanide 7 (2.0 mg, 1.5 × 10−5 mol) were dissolved in benzene-d6 in an NMR tube, 1H NMR spectrum of the mixture showed the existence of 2 and mono(isocyanide) complex 11 in 1 : 4 ratio. Addition of isocyanide 7 (2.0 mg, 1.5 × 10−5 mol) to the resulting solution afforded bis(isocyanide) complex 9 almost quantitatively as observed by 1H NMR spectroscopy.cis-Bis(dimethylphenylphosphine)dichloropalladium 14
To a benzene (10 cm3) solution of cis-bis(phenyl isocyanide)dichloropalladium17 (0.472 g, 1.23 mmol) in a Schlenk flask (50 cm3) was added dimethylphenylphosphine (0.4 cm3, 2.8 mmol). The red solution was stirred for 40 min and then the solvent was distilled off. Recrystallization of the resulted sticky solid from benzene afforded 14 as orange needles in the yield of 0.540 g (1.19 mmol, 97%).[η2-Tetrakis(tert-butyldimethylsilyl)disilene]bis(dimethylphenylphosphine)palladium 13
To a mixture of palladium dichloride 14 (0.102 g, 0.293 mmol) and 1,2-dilithiodisilane 5 (0.157 g, 0.296 mmol) in a degassed Schlenk flask (50 cm3) was introduced THF (5 ml) dried over K mirror through a vacuum line. The mixture was stirred for 2 h at rt and then THF was distilled off. Addition of toluene (40 cm3) to the yellow–brown residue and then the resulting precipitate was filtered off. Removal of toluene in vacuo and then recrystallization from toluene at −20 °C gave 13 (0.035 g, 0.04 mmol) in 20% yield. 13: mp 148 °C (decomp.); δH (400 MHz, C6D6) 0.49 (s, 12 H), 0.54 (s, 12 H), 1.25 (s, 36 H), 1.38 (d, 2JP–H 4.8 Hz, 12 H), 7.00 (dd, 4 H), 7.07 (d, 2 H), 7.47 (dd, 4 H); δC (100 MHz, THF-d8) 3.1, 3.3 (SiCH3), 20.1 (SiC(CH3)3), 20.8 (dd, 1JP–C 11 Hz, P(CH3)3), 30.2 (C(CH3)3), 128.9 (dd, 3JP–C = 4 Hz, 5JP–C = 2 Hz, CAr(m)), 129.4 (CAr(p)), 131.5 (dd, 2JP–C = 6 Hz, 4JP–C = 4 Hz, CAr(o)), 141.9 (dd, 1JP–C = 12 Hz, 2JP–C = 5 Hz, Cipso); δSi (79 MHz, C6D6) −44.8 (dd, 2JP(cis)–Si = 21 Hz, 2JP(trans)–Si = 80 Hz), +4.8; δP (161 MHz, C6D6) −19.0.Crystallography
Single crystals of complexes 8, 9 and 13 were obtained by recrystallization from toluene, mounted in Apiezon grease and transferred to the cold gas stream of the diffractometer. X-Ray data were collected on a Rigaku/MSC Mercury CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The data were corrected for Lorentz and polarization effects. The structures were solved by direct methods and refined by full-matrix least squares against F2 using all data (SHELXL-97).19In the single crystal of complex 8, two crystallographically independent molecules (molecule 1 and molecule 2) exists in an asymmetric unit. In molecule 1, one t-BuMe2Si group on Si(2) is disordered over two sites with occupancies 0.862(3) [central silicon atom: Si(6)] and 0.138(3) [central silicon atom: Si(13)]. All tert-butyl and methyl carbon atoms on minor Si(13) were refined with isotropic displacement parameters.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 12.998(4), b = 13.561(4), c = 16.667(5) Å, α = 97.695(2), β = 104.615(2), γ = 114.174(3)°, V = 2496.3(12) Å3, T = 150 K, Z = 2, µ(Mo-Kα) = 0.554 mm−1, 18243 reflections measured, 10160 unique (Rint = 0.0313) which were used in all calculations. Final R1 and wR2 were 0.0538 (I > 2σ(I)) and 0.1112 (all data), respectively.
(no. 2), a = 11.326(3), b = 11.988(3), c = 19.426(5) Å, α = 93.507(3), β = 94.393(3), γ = 100.588(4)°, V = 2577.5(11) Å3, T = 173 K, Z = 2, µ(Mo-Kα) = 0.586 mm−1, 19373 reflections measured, 10555 unique (Rint = 0.0329) which were used in all calculations. Final R1 and wR2 were 0.0503 (I > 2σ(I)) and 0.0882 (all data), respectively.
(no. 2), a = 12.998(4), b = 13.561(4), c = 16.667(5) Å, α = 97.695(2), β = 104.615(2), γ = 114.174(3)°, V = 2496.3(12) Å3, T = 150 K, Z = 2, µ(Mo-Kα) = 0.554 mm−1, 18243 reflections measured, 10160 unique (Rint = 0.0313) which were used in all calculations. Final R1 and wR2 were 0.0538 (I > 2σ(I)) and 0.1112 (all data), respectively.
(no. 2), a = 11.326(3), b = 11.988(3), c = 19.426(5) Å, α = 93.507(3), β = 94.393(3), γ = 100.588(4)°, V = 2577.5(11) Å3, T = 173 K, Z = 2, µ(Mo-Kα) = 0.586 mm−1, 19373 reflections measured, 10555 unique (Rint = 0.0329) which were used in all calculations. Final R1 and wR2 were 0.0503 (I > 2σ(I)) and 0.0882 (all data), respectively.
CCDC reference numbers 282912–282914.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512414j
Acknowledgements
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Grants-in-Aid for Scientific Research on Priority Areas (No. 14078203, “Reaction Control of Dynamic Complexes”) and on Scientific Research (A) (No. 16205007). We also thank Dow Corning Toray Co. Ltd for their generous gift of several silicon chemicals.References
- G. Frenking and W. Fröhlich, Chem. Rev., 2000, 100, 717 CrossRef CAS; F. R. Hartley, in Comprehensive Organometallic Chemistry, ed. G. Wilkinson, F. G. A. Stone and E. W. Abel, Pergamon Press, Oxford, 1982, vol. 6 Search PubMed.
- M. J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C71; J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939 RSC.
- Bent angle θ (degrees) defined here is similar to the pyramidalization angle defined for alkene complexes by Morokuma and Borden: (a) K. Morokuma and W. T. Borden, J. Am. Chem. Soc., 1991, 113, 1912 CrossRef CAS; The bent angle is related to bent-back angle (β) defined by Ibers and Stalick as θ = 90 − β; (b) J. K. Stalick and J. A. Ibers, J. Am. Chem. Soc., 1970, 92, 5333 CrossRef CAS; (c) S. D. Ittel and J. A. Ibers, Adv. Organomet. Chem., 1976, 14, 33 CAS.
- (a) E. K. Pham and R. West, J. Am. Chem. Soc., 1989, 111, 7667 CrossRef CAS; (b) E. K. Pham and R. West, Organometallics, 1990, 9, 1517 CrossRef CAS; (c) E. K. Pham and R. West, J. Am. Chem. Soc., 1996, 118, 7871 CrossRef CAS.
- (a) H. Hashimoto, Y. Sekiguchi, T. Iwamoto, C. Kabuto and M. Kira, Organometallics, 2002, 21, 454 CrossRef CAS; (b) H. Hashimoto, Yu. Sekiguchi, Yo. Sekiguchi, T. Iwamoto, C. Kabuto and M. Kira, Can. J. Chem., 2003, 81, 1241 CrossRef CAS; (c) M. Kira, T. Iwamoto, Y. Sekiguchi and C. Kabuto, J. Am. Chem. Soc., 2004, 126, 12778 CrossRef CAS.
- (a) D. H. Berry, J. H. Chey, H. S. Zipin and P. J. Carroll, J. Am. Chem. Soc., 1990, 112, 452 CrossRef CAS; (b) P. Hong, N. H. Damrauer, P. J. Carroll and D. H. Berry, Organometallics, 1993, 12, 3698 CrossRef CAS; D. H. Berry, J. H. Chey, H. S. Zipin and P. J. Carroll, Polyhedron, 1991, 10, 1189 CrossRef CAS.
- H. Hashimoto, K. Suzuki, W. Setaka, C. Kabuto and M. Kira, J. Am. Chem. Soc., 2004, 126, 13628 CrossRef CAS.
- R. Fischer, M. Zirngast, M. Flock, J. Baumgartner and C. Marschner, J. Am. Chem. Soc., 2005, 127, 70 CrossRef CAS.
- M. Kira, T. Iwamoto, D. Yin, T. Maruyama and H. Sakurai, Chem. Lett., 2001, 910 CrossRef CAS.
- M. Kira, T. Maruyama, C. Kabuto, K. Ebata and H. Sakurai, Angew. Chem., Int. Ed. Engl., 1994, 33, 1489 CrossRef; M. Kira, T. Iwamoto, T. Maruyama, T. Kuzuguchi, D. Yin, C. Kabuto and H. Sakurai, J. Chem. Soc., Dalton Trans., 2002, 1539 RSC.
- A digermene platinum complex was synthesized by a similar method: A. Castel, P. Riviere, J. Satge, D. Desor, M. Ahbala and C. Abdenadher, Inorg. Chim. Acta, 1993, 212, 51 Search PubMed.
- Unfortunately, no single crystals of 11 suitable for X-ray crystallography were obtained.
- M. Kaftory, M. Kapon and M. Botoshansky, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, Chichester, 1998, vol. 2, ch, 5, p. 181 Search PubMed.
- Y. Pan, J. T. Mague and M. H. Fink, Organometallics, 1992, 11, 3495 CrossRef CAS; M. Murakami, T. Yoshida and Y. Ito, Organometallics, 1994, 13, 2900 CrossRef CAS; M. Suginome, H. Oike, S.-S. Park and Y. Ito, Bull. Chem. Soc. Jpn., 1996, 69, 289 CAS.
- We assume that the avaraged bent angles (θav.) are taken as indices of the mean strength of σ-donor abilities of two residual ligands.
- All calculations were carried out at the B3LYP level using a Gaussian 98 program, revision A.11.4; Gaussian, Inc.: Pittsburgh, PA, 2001. The basis sets were 6-31G(d) for H, C, Si, P atoms and Lanl2dz for Pd atom.
- Theoretical calculations for related disilene complexes with methyl isocyanide ligands 8″ and 9″ showed no serious differences in the effects between pheny isocyanide and methyl isocyanide as ligands. See the ESI† for the details of the calculations.
- J. R. Doyle, P. E. Slade and H. B. Jonassen, Inorg. Synth., 1960, 6, 218.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data for DFT calculations for model disilene complexes 8′, 8″, 9′ and 9″. See DOI: 10.1039/b512414j |
| This journal is © The Royal Society of Chemistry 2006 |