A novel multidecker sandwich complex from the reaction of ferrocene with GaCl3
Stefan Scholza, Jennifer C. Greenb, Hans-Wolfram Lernera, Michael Boltec and Matthias Wagner*a
aInstitut für Anorganische Chemie, J. W. Goethe-Universität Frankfurt, Marie-Curie-Str. 11, D-60439, Frankfurt (Main), Germany.. E-mail: Matthias.Wagner@chemie.uni-frankfurt.de
bInorganic Chemistry Laboratory, South Parks Road, Oxford, UK OX1 3QR
cInstitut für Organische Chemie, J. W. Goethe-Universität Frankfurt, Marie-Curie-Str. 11, D-60439, Frankfurt (Main), Germany
First published on 13th December 2001
Abstract
A redox reaction takes place between GaCl3 and an excess of ferrocene, leading to the multidecker sandwich complex 1, [–Ga(C5H5)Fe(C5H5)Ga(C 5H5)Fe(C5H5)–]n[GaCl4]2n, which features an array of alternating Ga(I) and Fe(II) ions bridged by cyclopentadienyl moieties.
Transition metals as well as main group elements form a wide variety of complexes with the cyclopentadienyl ligand.1,2 Many of these compounds have been investigated by X-ray crystallography. Two species are particularly noteworthy in the context of this paper: (i) ferrocene, (η5-C5H5)2Fe,3 the bis(cyclopentadienyl) complex of Fe(II) and (ii) the pentamethylcyclopentadienyl complex of Ga(I), which forms a hexameric cluster (η5-C5Me5Ga)64 in the solid state. Related, but less stable π-complexes of main group elements with uncharged aromatic systems are also known. Schmidbaur et al., for example, succeeded in the synthesis and structural characterisation of various Ga(I) arene complexes.5,6
A landmark in the field was the discovery of multidecker sandwich complexes (e.g. of nickel and rhodium) with the 2,3-dihydro-1,3-diborolyl ligand, which is able to coordinate two transition metal atoms in an η5 fashion, thereby adopting a bridging position.7 Analogous compounds consisting of a main group element and a transition metal are, however, very rare {cf. (C5H5)Co(C2Me2B2Me2C)Tl8 and [(C5H5)Co(C2Et2B2Me2C)]2Sn9}.
Herein we report on the polymeric multidecker sandwich complex 1 featuring an array of alternating Ga(I) and Fe(II) ions bridged by cyclopentadienyl moieties (Scheme 1). When an equimolar solid mixture of ferrocene and GaCl3 is treated with benzene,† a blue–green solution is obtained, which contains paramagnetic species (1H NMR spectroscopic monitoring). From this solution, a first crop of blue–green crystals was isolated and identified as [(C5H5)2Fe][GaCl4] by means of X-ray crystallography.10 The mother-liquor was evaporated and the pulpy residue recrystallised from CH2Cl2 to give a second crop consisting of red platelets, which are highly sensitive to air and moisture. An X-ray crystal structure determination‡ of the material revealed the multidecker sandwich complex 1 featuring Ga(I) centers, each coordinated to two different ferrocene molecules (Scheme 1). This leads to the conclusion that a redox process has taken place [Scheme 1, eqn. 1(a)] in the system ferrocene/GaCl3, rather than an electrophilic substitution of the Cp ring as in the case of the related system ferrocene/BBr311 [Scheme 1, eqn. 1(b)].
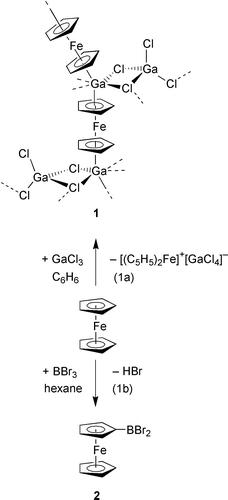 | ||
| Scheme 1 Reactions of EX3 (E = B, Ga; X = Cl, Br) with ferrocene. | ||
When a smaller amount of benzene is employed, we observe the immediate precipitation of microcrystalline [(C5H5)2Fe][GaCl4], as well as the formation of two separate liquid phases. A similar phenomenon has been reported by Schmidbaur et al.12 upon dissolving Ga[GaBr4] in aromatic solvents and has been attributed to the presence of solvent clathrates at higher concentrations.
GaCl3 as well as GaCl3·dioxane, can be reduced not only with ferrocene, but also with decamethylferrocene and cobaltocene. The corresponding oxidation products, [(C5Me5)2Fe][GaCl4] and [(C5H5)2Co][GaCl4], have been identified by X-ray crystal structure analysis.13
A comparison of the 1H NMR spectra (d6-benzene) of 1 (4.38 ppm; h1/2 20.4 Hz) and ferrocene (4.00 ppm) reveals a downfield shift of 0.38 ppm for the former. The Ga(I) ion of Ga[GaCl4] exhibits a resonance at δ −715 in its 71Ga NMR spectrum (d6-benzene), which is shifted by 45 ppm to lower field (δ −670) when ferrocene is added to the solution. This leads to the conclusion that a certain proportion of the Ga(I) centers remain coordinated to the electron-rich ferrocene molecules, even in the presence of an excess of benzene. Compound 1 crystallises in the monoclinic space group P21/n (Fig. 1).‡
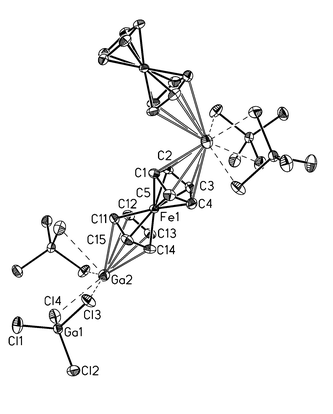 | ||
| Fig. 1 Solid state structure of 1. H atoms have been omitted for clarity. Selected bond lengths (pm) and angles (°): Ga(2)–C(1) 299.8(3), Ga(2)–C(2) 301.5(3), Ga(2)–C(3) 312.0(3), Ga(2)–C(4) 315.4(3), Ga(2)–C(5) 308.4(3), Ga(2)–C(11) 334.6(3), Ga(2)–C(12) 331.5(3), Ga(2)–C(13) 316.6(3), Ga(2)–C(14) 311.3(3), Ga(2)–C(15) 322.4(3), Ga(2)–Cl(2) 338.0(1), Ga(2)–Cl(3) 314.9(1), Ga(2a)–Cl(3) 326.6(1); Cl(1)–Ga(1)–Cl(2) 110.77(3), Cl(1)–Ga(1)–Cl(4) 109.97(3), Cl(2)–Ga(1)–Cl(4) 111.60(3), Cl(1)–Ga(1)–Cl(3) 109.81(3), Cl(2)–Ga(1)–Cl(3) 107.49(3), Cl(4)–Ga(1)–Cl(3) 107.10(3); mean value Ga(2)–COG(Cp) 291.2. | ||
The Ga(I) centers are coordinated by two chelating [GaCl4]− moieties and two cyclopentadienyl ligands. Each [GaCl4]− anion links two Ga(I) centres, thereby creating polymeric rods running through the crystal lattice. Perpendicular to these rods, zigzag chains consisting of Ga(I) ions and ferrocene molecules are established. Each Ga(I) centre is coordinated to two Cp rings of two different ferrocene molecules in an η5 fashion. The mean distance Ga(2)–C(Cp) between Ga(I) and the Cp carbon atoms is 315.3 pm, a value very close to the mean Ga–C(benzene) distance of 316 pm observed in the complex [Ga(C6H6)2]+[GaCl4]− .5 Compared to (η5-C5Me5Ga)6, which features a mean Ga–C(Cp*) distance of 240.0 pm,4 the Ga(2)–C(Cp) bond lengths in 1 are elongated by 75.3 pm. An average distance of 291.2 pm is found between Ga(2) and the centres of gravity (COG) of the cyclopentadienyl rings in compound 1 {cf. [Ga(C6H6)2]+[GaCl4] −: average distance Ga–COG(C6H6) 284.7 pm5}. The [Fe(C5H5)2]2Ga fragment is bent by an angle Cp(COG)–Ga(2)–Cp(COG) of 110.3°.
To gain further insight into the relative stability of Ga(I)–benzene vs. Ga(I)–ferrocene complexes, DFT calculations were performed.§ The optimised structure of [Fe(C5H5)2]2Ga+ had an average Ga–C distance of 293 pm and an angle Cp(COG)–Ga(2)–Cp(COG) of 158°. The optimised angle for the unit is sufficiently different from that found in the crystal structure to suggest that the coordination of the Ga(I) to the [GaCl4]− units partly determines the angle. The optimisation was repeated with the angle fixed at 110° as found experimentally; the average Ga–C distance lengthened to 305 pm, closer to that found experimentally. The energy of the species with an angle of 110° is 22 kJ mol−1 above the ground state species, indicating a soft surface for bending. The binding energy of two ferrocene molecules to Ga+ was calculated as 213 kJ mol−1 while that for formation of [Ga(C6H6)2]+ from two benzene molecules and Ga+ is calculated to be less at 136 kJ mol−1. Fragment analysis of [Fe(C5H5)2]2Ga+ indicated a transfer of 0.4 electrons into the gallium 4p orbitals compared with a zero occupancy for Ga+. This points towards a small covalent interaction between the ferrocenes and the Ga(I) ion.
This research was supported by the ‘Deutsche Forschungsgemeinschaft’ (DFG).
Notes and references
- N. J. Long, Metallocenes, Blackwell Science, London, 1998. Search PubMed.
- P. Jutzi and N. Burford, Chem. Rev., 1999, 99, 969 CrossRef CAS.
- Ferrocenes, ed. A. Togni and T. Hayashi, VCH, Weinheim, 1995. Search PubMed.
- D. Loos, E. Baum, A. Ecker, H. Schnöckel and A. J. Downs, Angew. Chem., Int. Ed. Engl., 1997, 36, 860 CrossRef CAS.
- H. Schmidbaur, U. Thewalt and T. Zafiropoulos, Organometallics, 1983, 2, 1550 CrossRef CAS.
- H. Schmidbaur, W. Bublak, B. Huber and G. Müller, Organometallics, 1986, 5, 1647 CrossRef CAS.
- W. Siebert, Angew. Chem., Int. Ed. Engl., 1985, 24, 943 CrossRef.
- K. Stumpf, H. Pritzkow and W. Siebert, Angew. Chem., Int. Ed. Engl., 1985, 24, 71 CrossRef.
- H. Wadepohl, H. Pritzkow and W. Siebert, Organometallics, 1983, 2, 1899 CrossRef CAS.
- Crystal data for [(C5H5)2Fe][GaCl4]: C10H10Cl4FeGa, orthorhombic, space group Pnma, a = 1371.7(1), b = 860.0(1), c = 1201.4(8) pm. CCDC reference number 170258..
- T. Renk, W. Ruf and W. Siebert, J. Organomet. Chem., 1976, 120, 1 CrossRef CAS.
- M. Uson-Finkenzeller, W. Bublak, B. Huber, G. Müller and H. Schmidbaur, Z. Naturforsch. Teil B, 1986, 41, 346 Search PubMed.
- Crystal data for [(C5Me5)2Fe][GaCl4]: C20H30Cl4FeGa, orthorhombic, space group Cccm, a = 1252.95(2), b = 2276.16(3), c = 1985.72(2) pm unpublished work. Crystal data for [(C5H5)2Co][GaCl4]·C6H6: C16H16Cl4CoGa, monoclinic, space group Cc, a = 1174.4(1), b = 1522.6(1), c = 1113.5(1) pm, β = 106.851(5)°. CCDC 153730.
- E. J. Baerends, A. Berces, C. Bo, P. M. Boerringter, L. Cavallo, L. Deng, R. M. Dickson, D. E. Ellis, L. Fan, T. H. Fischer, C. Fonseca Guerra, S. J. van Gisbergen, J. A. Groeneveld, O. V. Gritsenko, F. E. Harris, P. van den Hoek, H. Jacobsen, G. van Kessel, F. Kootstra, E. van Lenthe, V. P. Osinga, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, P. Ros, P. R. T. Schipper, G. Schreckenbach, J. G. Snijiders, M. Sola, D. Swerhone, G. te Velde, P. Vernooijis, L. Versluis, O. Visser, E. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo and T. Ziegler, ADF Program System Release 1999, 1999, 5 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 2398 CrossRef.
- (a) J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef; (b) J. P. Perdew, Phys. Rev. B, 1986, 34, 7046.
Footnotes |
| † All manipulations were carried out under anaerobic and anhydrous conditions; in a typical experiment 40 mg (0.21 mmol) of ferrocene and 37 mg (0.21 mmol) of GaCl3 were used. Yields of 1 vary between 40 and 65% (relative to the maximum yield obtainable from the reaction of 3 equiv. of ferrocene and 4 equiv. of GaCl3) and are dependent from the time allowed for crystallisation; formation of [(C5H5)2Fe][GaCl4] is generally quantitative. |
| ‡ Crystal data for 1: C10H10Cl4FeGa2, M = 467.27, monoclinic, space group P21/n, a = 1056.89(1), b = 1455.58(1), c = 1075.24(1) pm, β = 113.243(1)°, V = 1519.89(2) 106 pm3, T = 173(2) K, Z = 4, μ(Mo-Kα) = 5.138 mm−1, 31849 reflections measured, 3612 unique (Rint = 0.0477) which were used in all calculations. The final wR(F2) was 0.0575 (all data). CCDC reference number 153729. See http://www.rsc.org/suppdata/cc/b1/b108515h/ for crystallographic data in cif or other electronic format. |
| § Density functional calculations were carried out using the Amsterdam Density Functional code (ADF 2000.2).14 The basis set used triple-ζ accuracy sets of Slater type orbitals, with relativistic corrections, with a single polarisation function added to the main group atoms. The cores of the atoms were frozen [C (1s), Ga (2p), Fe(2p)]. The GGA (non-local) method was used, using Vosko, Wilk and Nusair’s local exchange correlation15 with non-local-exchange corrections by Becke16 and non-local correlation corrections by Perdew.17 Fragment analyses were carried out on {Ga[Fe(C5H5)2]2} +. This involved single point calculations using the MOs of the gallium and ferrocene as fragments as the basis set for the molecular calculations. The fragments retain the geometries found for the optimised molecular structure. |
| This journal is © The Royal Society of Chemistry 2002 |