Asymmetric deprotonation and dearomatising cyclisation of N-benzyl benzamides using chiral lithium amides: formal synthesis of (–)-kainic acid
Jonathan
Clayden
*a,
Christel J.
Menet
a and
Darren J.
Mansfield
b
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, UK M20 5LG. E-mail: j.p.clayden@man.ac.uk
bAventis CropScience SA, Centre de Recherche de La Dargoire, 14-20 rue Pierre Baizet - BP 9163, 69263, Lyon Cedex 09, France
First published on 3rd December 2001
Abstract
Chiral lithium amides deprotonate N-benzyl benzamides enantioselectively, initiating an asymmetric dearomatising cyclisation to enantiomerically enriched isoindolinones.
We have previously reported1 that lithiated N-benzyl amides cyclise at temperatures approaching 0 °C (−78 °C in the presence of HMPA2,3) to yield dearomatised products which take the form of partially saturated isoindolinones (Scheme 1). These heterocycles, with their three new stereogenic centres and versatile functional groups, are versatile synthetic intermediates, and we have published a synthesis of (±)-kainic acid using the cyclisation of 1b as a key step.4 In this paper we report an asymmetric cyclisation achieved† by means of chiral lithium amide bases which proceeds via an asymmetric benzylic deprotonation.
![Dearomatising cyclisation of lithiated N-benzyl benzamides. (i) [for (±)-5] LDA, −78–+25 °C, 1.5 h; (ii) [for (−)-5] chiral lithium amide, −78–+25 °C, 1.5 h; (iii) NH4Cl; (iv) HCl, H2O.](/image/article/2002/CC/b109188c/b109188c-s1.gif) | ||
| Scheme 1 Dearomatising cyclisation of lithiated N-benzyl benzamides. (i) [for (±)-5] LDA, −78–+25 °C, 1.5 h; (ii) [for (−)-5] chiral lithium amide, −78–+25 °C, 1.5 h; (iii) NH4Cl; (iv) HCl, H2O. | ||
α-Amido benzyllithiums related to 2 may be generated enantioselectively with s-BuLi–(−)-sparteine and are configurationally stable over a period of 10 h at −78 °C.5,6 However, while treatment of 1a with s-BuLi–(−)-sparteine7 in THF gave the cyclised product 5a, it turned to be racemic. Insufficient solubility prevented the use of Et2O or t-BuOMe; toluene turned out to be too acidic, becoming lithiated itself and leading to benzyl ketones; no reactions took place in cumene.
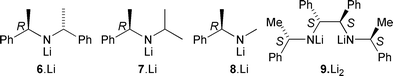
As we knew that LDA deprotonates 1a at temperatures approaching −40 °C,1 we turned to chiral versions of LDA, namely the lithium amides 6·Li, 7·Li, 8·Li and 9·Li2.8 Each base was used with the amide 1a, and the crude product was hydrolysed to the enone with aqueous hydrochloric acid. The products 5a–d were purified by flash chromatography, and the yields and enantiomeric excesses obtained are shown in Table 1. The best results were with 7·Li: not only did it give the highest enantiomeric excesses, but it was also the easiest to remove from the product mixture by a simple acid wash.‡ A single recrystallisation was enough to return the products 1a and 1b in >99% ee. Attempts to improve enantioselectivity by changing the temperature régime (entries 2, 10) were unsuccessful. The absolute stereochemistry of the products (which were all laevorotatory with 6·Li–8·Li and dextrorotatory with 9·Li2) was determined by X-ray crystallography of the enone (−)-10 obtained by bromination (Br2, Et3N, CH2Cl2: 80%) of enantiomerically pure (−)-5a.
| Entry | Starting material | Base | (−)-5 yield (%) | (−)-5 eea (%) | (−)-5 eeb (%) |
|---|---|---|---|---|---|
| a Ee after flash chromatography. b Ee after recrystallisation. c Reaction carried out −78–−60 °C, 16 h. d (+)-5 is the major enantiomer. e Reaction carried out −50 °C–+25 °C. | |||||
| 1 | 1a | 6 ·Li | 73 | 80 | >99 |
| 2 | 1a | 6 ·Lic | 65 | 50 | — |
| 3 | 1a | 7 ·Li | 72 | 80 | >99 |
| 4 | 1a | 8 ·Li | 72 | 17 | — |
| 5 | 1a | 9 ·Li2 | 38 | −75d | — |
| 6 | 1b | 6 ·Li | 64 | 75 | >99 |
| 7 | 1b | 7 ·Li | 72 | 81 | >99 |
| 8 | 1b | 9 ·Li2 | 23 | −30d | — |
| 9 | 1c | 6 ·Li | 59 | 73 | – |
| 10 | 1c | 6 ·Lie | 67 | 60 | — |
| 11 | 1d | 7 ·Li | 87 | 84 | — |
| 12 | 11 | 7 ·Li | 70 | 60 | 0 |
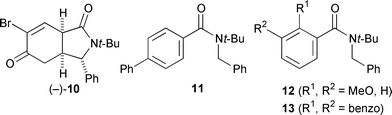
Enantioselective cyclisation of the para-substituted amide 11 was also successful under these conditions, though unfortunately it was impossible to improve the enantiomeric excess of the product by recrystallisation. However, amides bearing ortho substituents such as 12 or 13 gave either low yields or low enantioselectivities, probably because such ortho-substituted tertiary aromatic amides are chiral compounds at low temperatures.§
As for the origin of the enantioselectivity, two limiting possibilities present themselves. The first is that the reaction proceeds via an asymmetric deprotonation of the achiral starting material 1, generating an enantiomerically enriched organolithium which is configurationally stable on the timescale of the reaction and which cyclises stereospecifically. The second is that the reaction is a stereoselective cyclisation of an intermediate organolithium (which may or may not be configurationally stable) under the asymmetric influence of the amine such as 7·H regenerated after deprotonation. The known configurational stability of lithio amides similar to 25,6 at least does not rule out the first mechanism. To distinguish between these two possibilities, we carried out two experiments.9
In the first experiment, we treated 1a with t-BuLi to generate organolithium (±)-2a, and then added chiral amine 7·H, recreating the conditions of the asymmetric cyclisation but with a starting organolithium that must, at least initially, be racemic (Scheme 2). After an aqueous quench and acidic work-up, the product 5a of this reaction was nearly racemic, proving that the enantioselectivity in the asymmetric reaction is determined before the cyclisation step.
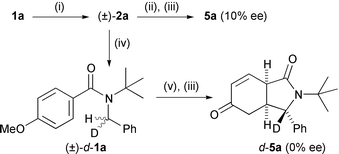 | ||
| Scheme 2 Mechanism of the cyclisation (i) t-BuLi, THF, −78 °C; (ii) 6·H, −78 °C–+25 °C; (iii) NH4Cl then HCl, H2O; (iv) CD3OD; (v) 6.Li, THF, −78–+25 °C. | ||
In the second experiment, we used racemic, deuterium-labelled (±)-d-1a (obtained by deuteration of (±)-2a, Scheme 2) as the starting material for the asymmetric cyclisation. Cyclisation of (±)-d-1a with 6·Li gave racemic material 5a which was >99% deuterated — a result consistent only with a primary kinetic isotope effect kH/kD of at least 100 which has completely overturned the enantioselectivity of the chiral base.10,11 This in turn implies the enantioselective step must be a step subject to a primary kinetic isotope effect, i.e. the deprotonation.
While the s-BuLi–(−)-sparteine has commonly been used for the enantioselective removal of one of a pair of enantiotopic geminal H atoms to give a chiral organolithium,7 the less basic chiral lithium amide bases are much more frequently employed to generate chiral planar anions, such as enolates, by selecting between enantiotopic enolisation sites of prochiral carbonyl compounds.8 By carrying out the asymmetric deprotonation at temperatures too low for cyclisation to occur (<−40 °C), we hoped to observe the asymmetric formation and external quench of a chiral, non-planar organolithium generated with a chiral lithium amide, a reaction observed before only with Cr(CO)3-complexed benzyl groups.12,13 When we treated the amide 1a with 7·Li at −78 °C, warmed to −40 °C over 1 h to ensure deprotonation, and then added an electrophile, the ee of the product was dependent on the electrophile used (Scheme 3). Presumably the rate of racemisation of the organolithium 2a is similar to the rate of reaction with an electrophile, and racemisation outpaces attack on MeI but not on CO2.¶ We conclude that asymmetric deprotonation of one of the pair of enantiotopic protons of 1 gives a benzylic organolithium with sufficient configurational stability to cyclise stereospecifically but insufficient configurational stability to prevent partial or complete racemisation on the timescale of its addition to an external electrophile.
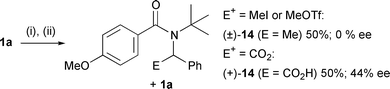 | ||
| Scheme 3 External electrophiles (i) 7·Li, −78–−40 °C, 1 h; (ii) E+. | ||
Our published route to (±)-kainic acid4 uses 18 as an intermediate. As a demonstration of the utility of the asymmetric cyclisation, we therefore converted 5d (which was produced in 84% ee by the most enantioselective cyclisation we could find, Table 1, entry 11) to 18 in four steps (Scheme 4). 5d could not be recrystallised to enantiomeric purity, but after addition of Me2CuLi and deprotection/hydrolysis of the enol ether 15, recrystallisation of pyrrolidinone 16 gave enantiomerically pure material in an overall yield of 48% from 5d. Boc protection of 16 and oxidation of the m-MeOC6H4 ring of 18 gave the enantiomerically pure ester 18, which we have converted to kainic acid in five steps.4
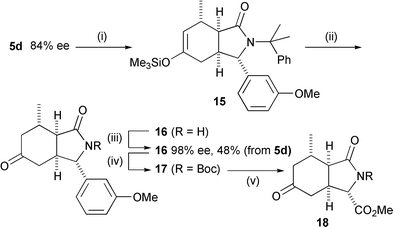 | ||
| Scheme 4 Formal synthesis of (−)-kainic acid (i) Me2CuLi, Me3SiCl; (ii) CF3CO2H; (iii) recrystallise; (iv) Boc2O, DMAP, Et3N, CH2Cl2; (v) RuCl3, NaIO4, H2O, MeCN then CH2N2. | ||
We are grateful to the EPSRC for a studentship (to CJM), to Dr C. S. Frampton for determining the absolute stereochemistry of 10, and to Miss K. Hebditch for studying the cyclisation of 12.
Notes and references
- J. Clayden, C. J. Menet and D. Mansfield, Org. Lett., 2000, 2, 4229 CrossRef CAS.
- A. Ahmed, J. Clayden and S. A. Yasin, Chem. Commun., 1999, 231 RSC.
- J. Clayden, K. Tchabanenko, S. A. Yasin and M. D. Turnbull, Synlett, 2001, 302 CAS.
- J. Clayden and K. Tchabanenko, Chem. Commun., 2000, 317 RSC.
- S. Wu, S. Lee and P. Beak, J. Am. Chem. Soc., 1996, 118, 715 CrossRef CAS; Y. S. Park, M. L. Boys and P. Beak, J. Am. Chem. Soc., 1996, 118, 3757 CrossRef CAS.
- M. Schlosser and D. Limat, J. Am. Chem. Soc., 1995, 117, 12342 CrossRef CAS; C. Barberis, N. Voyer, J. Roby, S. Chénard, M. Tremblay and P. Labrie, Tetrahedron, 2001, 57, 2965 CrossRef CAS.
- D. Hoppe and T. Hense, Angew. Chem., Int. Ed., 1997, 36, 2282 CrossRef CAS.
- P. O′Brien, J. Chem. Soc., Perkin Trans. 1, 1998, 1439 RSC.
- For a similar discussion, see: P. Beak, A. Basu, D. J. Gallagher, Y. S. Park and S. Thayumanavan, Acc. Chem. Res., 1996, 29, 552 Search PubMed.
- D. Hoppe, M. Paetow and F. Hintze, Angew. Chem., Int. Ed., 1993, 32, 394 CrossRef.
- D. R. Anderson, N. C. Faibish and P. Beak, J. Am. Chem. Soc., 1999, 121, 7553 CrossRef CAS.
- E. L. M. Cowton, S. E. Gibson (née Thomas), M. J. Schneider and M. H. Smith, Chem. Commun., 1996, 839 RSC.
- R. A. Ewin, A. M. MacLeod, D. A. Price, N. S. Simpkins and A. P. Watt, J. Chem. Soc., Perkin Trans. 1, 1997, 401 RSC.
- A. Ahmed, J. Clayden and M. Rowley, Chem. Commun., 1998, 297 RSC.
- A. Ahmed, R. A. Bragg, J. Clayden and K. Tchabanenko, Tetrahedron Lett., 2001, 42, 3407 CrossRef CAS.
- R. A. Bragg, J. Clayden, M. Bladon and O. Ichihara, Tetrahedron Lett., 2001, 42, 3411 CrossRef CAS.
- R. A. Bragg and J. Clayden, Tetrahedron Lett., 1999, 40, 8323 CrossRef CAS.
- F. Hammerschmidt and A. Hanninger, Chem. Ber., 1995, 128, 823 CAS.
- A. Ahmed, R. A. Bragg, J. Clayden, L. W. Lai, C. McCarthy, J. H. Pink, N. Westlund and S. A. Yasin, Tetrahedron, 1998, 54, 13277 CrossRef CAS; J. Clayden and J. H. Pink, Tetrahedron Lett., 1997, 38, 2561 CrossRef CAS.
- R. A. Bragg and J. Clayden, Tetrahedron Lett., 1999, 40, 8327 CrossRef CAS.
Footnotes |
| † Related cyclisations of N-benzyl-1-naphthamides14,15 have been made asymmetric by the use of a chiral auxiliary16 or chiral starting material.17 |
| ‡ 7·H was made in 61% yield by stirring (S)-α-methylbenzylamine with isopropyl bromide.18 |
| § Tertiary 1-naphthamides and ortho-substituted benzamides exhibit atropisomerism at low temperature,19 and hence 12 and 13 must be viewed as racemic compounds under the conditions of the deprotonation. This feature has a governing role in the cyclisation of chiral 1-naphthamides.20 |
| ¶ The absolute stereochemistry of (+)-14 (E = CO2H) is unknown. |
| This journal is © The Royal Society of Chemistry 2002 |