Parasite glycoconjugates. Part 11.1 Preparation of phosphodisaccharide synthetic probes, substrate analogues for the elongating α-D-mannopyranosylphosphate transferase in the Leishmania
Andrew J.
Ross
a,
Irina A.
Ivanova
a,
Michael A. J.
Ferguson
b and
Andrei V.
Nikolaev
*a
aDepartment of Chemistry, University of Dundee, Dundee, UK DD1 4HN
bDepartment of Biochemistry, University of Dundee, Dundee, UK DD1 4HN
First published on 11th December 2000
Abstract
A set of phosphodisaccharides, substrate analogues, which will be used to study acceptor–substrate specificity of the Leishmania biosynthetic enzymes, are synthesized using the Koenigs–Knorr and trichloroacetimidate methods for the glycosylation reactions, SN2 nucleophilic displacement of a triflic ester for epimerization, and the glycosyl hydrogenphosphonate method for phosphorylation.
Introduction
Throughout the tropics and subtropics Leishmania parasites cause a variety of diseases ranging from self-limiting skin lesions to the often fatal visceral leishmaniasis. The surface lipophosphoglycan (LPG) produced by the infectious promastigote stage of all species of the Leishmania contains a polymeric section consisting of (1→6)-linked β-D-galactosyl-(1→4)-α-D-mannosyl phosphate repeating units. The importance of the LPG for parasite infectivity and survival![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 2 makes the enzymes responsible for the biosynthesis of this glycoconjugate of great interest. Phospho-oligosaccharide fragments of the LPG of L. donovani, L. mexicana and L. major were synthesized3–6 in our laboratory and tested as acceptor substrates (in vitro) for the Leishmania
α-D-mannopyranosylphosphate transferase (MPT) responsible for the transfer of α-D-Manp phosphate from GDP-Man to the growing phosphoglycan chain. It was shown7 that the phosphodisaccharide 14,8 (representing one repeating unit of the phosphoglycan) is the minimal structure exhibiting acceptor substrate activity for the MPT.
2 makes the enzymes responsible for the biosynthesis of this glycoconjugate of great interest. Phospho-oligosaccharide fragments of the LPG of L. donovani, L. mexicana and L. major were synthesized3–6 in our laboratory and tested as acceptor substrates (in vitro) for the Leishmania
α-D-mannopyranosylphosphate transferase (MPT) responsible for the transfer of α-D-Manp phosphate from GDP-Man to the growing phosphoglycan chain. It was shown7 that the phosphodisaccharide 14,8 (representing one repeating unit of the phosphoglycan) is the minimal structure exhibiting acceptor substrate activity for the MPT.
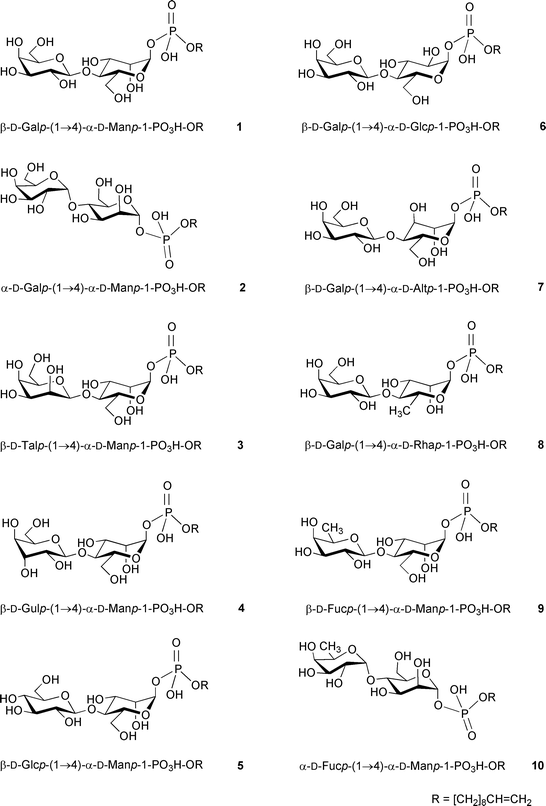
In Part 98 of this series, we disclosed our interest in the design and synthesis of various structural analogues of compound 1 to test the fine acceptor substrate specificity of the MPT and to gain more information about enzyme–substrate recognition. Thus, phosphodisaccharides 2–5, which are epimers of the substrate 1 at C-1′, C-2′, C-3′ or C-4′, respectively, have been synthesized.
We now report the chemical synthesis of the disaccharide phosphates 6–10. Compounds 6 and 7 are epimers of the substrate 1 at C-2 and C-3, respectively, of the D-mannopyranose moiety. Compounds 8 and 9 are substrate analogues deoxygenated at positions C-6 and C-6′, respectively. In this context, the preparation of the analogue 10, which is an epimer of compound 9 at C-1′ and could be (as well as the analogue 9 itself) a potential inhibitor of the enzyme, is also described. The information obtained from testing the acceptor activity of the substrate analogues 2–10 will be used to predict which sugar hydroxy groups of compound 1 are involved in enzyme–substrate recognition events and to design potential enzyme inhibitors.
Results and discussion
The synthetic schemes for the preparation of the phosphodisaccharides 6–10 consist of a few general steps (Scheme 1): 1) synthesis of fully protected disaccharide derivatives A; 2) anomeric de-O-protection ( → B); 3) H-phosphonylation at position O-1 ( → C); 4) coupling of the H-phosphonates C with dec-9-en-1–ol (using the glycosyl H-phosphonate method)9 to furnish the protected glycosyl phosphodiesters D; 5) total de-O-protection.
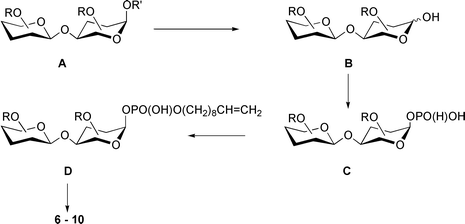 | ||
| Scheme 1 R = Ac, or Bz; R′ = Ac, or Bz, or Bn. | ||
The octa-O-acetyl-α,β-lactose 11 (α∶β = 7∶1; which is a precursor of the phosphodisaccharide 6; Scheme 2) was prepared by conventional acetylation of α-lactose and then converted to the hemiacetal 12 (83%; α∶β = 4∶1) by anomeric de-O-acylation3–6,8–10 with dimethylamine in CH3CN–THF. H-Phosphonylation3–6,8–10 of compound 12 with triimidazolylphosphine (prepared in situ from PCl3, imidazole and Et3N) followed by mild hydrolysis produced a mixture of α- and β-linked H-phosphonates 13 (α∶β = 4∶1, as evinced from 1H and 31P NMR spectra, see Experimental section), which were not separable by flash-column chromatography. This mixture was converted to the pure α-(H-phosphonate) 14 (48% based on the hemiacetal 12) by treatment with H3PO3 in acetonitrile. This procedure was developed first for the preparation10 of pure 2,3,4,6-tetra-O-benzoyl-α-D-galactopyranosyl H-phosphonate and utilizes the higher reactivity of the β-linked glycosyl H-phosphonate, converting it to either the α-linked isomer (as a result of SN2-attack), or easily separable hemiacetal derivative (product of acid-catalyzed cleavage of the H-phosphonate group).
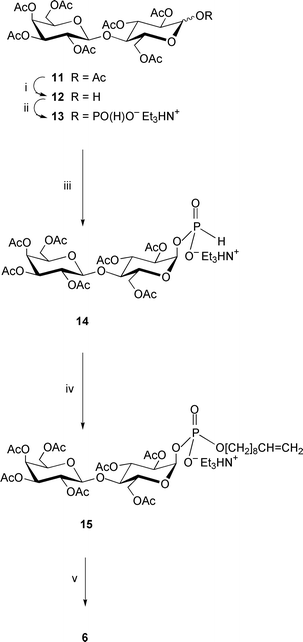 | ||
| Scheme 2 Reagents: i, Me2NH, MeCN–THF; ii, (a) triimidazolylphosphine, MeCN; (b) Et3NHHCO3, water (pH 7); iii, H3PO3, MeCN; iv, (a) dec-9-en-1-ol, trimethylacetyl chloride, pyridine; (b) I2, pyridine–water; v, NaOMe, MeOH. | ||
Synthesis of the protected benzyl β-D-galactopyranosyl-(1→4)-α-D-altropyranoside 22 (which is a precursor of the phosphodisaccharide 7; Scheme 3) was performed from benzyl 2,3,6-tri-O-benzoyl-α-D-altropyranoside 20 and acetobromogalactose 21. The altropyranoside 20 in turn was prepared starting from benzyl α-D-mannopyranoside, which was converted first to the 2-O-benzoate 17 (65%) by 4,6-O-isopropylidenation11 with 2-methoxypropene ( → 16) followed by selective benzoylation12 with benzoyl cyanide in the presence of Et3N. Successive reaction with triflic anhydride in CH2Cl2 in the presence of pyridine led to the triflate 18, which reacted with tetrabutylammonium benzoate (Bu4NOBz) in toluene (60 °C) to give the altroside 19 (73%). The D-altro-configuration of the derivative 19 was confirmed by the characteristic values of J2,3 = J3,4 = 3.0 Hz in 1H NMR spectrum. Further, compound 19 was converted to the glycosyl acceptor 20 (67%) by acid hydrolysis followed by selective 6-O-benzoylation with benzoyl cyanide.
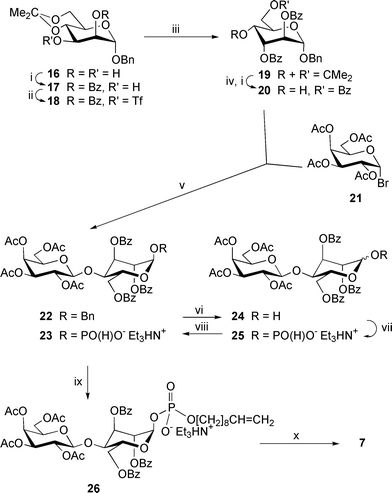 | ||
| Scheme 3 Reagents: i, BzCN, Et3N, MeCN; ii, Tf2O, CH2Cl2–pyridine; iii, Bu4NOBz, toluene; iv, 80% AcOH; v, AgOTf, Ag2CO3, MS 4 Å, CH2Cl2; vi, H2, Pd(OH)2/C, THF; vii, (a) triimidazolylphosphine, MeCN; (b) Et3NHHCO3, water (pH 7); viii, H3PO3, MeCN; ix, (a) dec-9-en-1-ol, trimethylacetyl chloride, pyridine; (b) I2, pyridine–water; x, NaOMe, MeOH. | ||
Glycosylation of the acceptor 20 with the bromide 21 in the presence of silver triflate (AgOTf), silver carbonate and molecular sieves 4 Å in dichloromethane provided the disaccharide 22 in 52% yield. Hydrogenolysis of compound 22 over Pd(OH)2/C afforded a mixture of α- and β-hemiacetals 24 in the ratio α∶β = 0.8∶1 (confirmed by 1H NMR data, see Experimental section). Probably, the mutarotation was facilitated because of unfavourable 1,3-synaxial interaction between 1-OH and 3-benzoate in the α-hemiacetal. The anomeric mixture 24 was converted to the pure α-(H-phosphonate) 23 using the same procedure as described for the H-phosphonate 14: i.e., the reaction with triimidazolylphosphine and mild hydrolysis ( → 25) followed by treatment with H3PO3 in CH3CN. This produced the H-phosphonate 23 (35% based on the disaccharide 22) along with the recovered hemiacetal 24 (49%).
The hepta-O-acetyl-β-D-galactopyranosyl-(1→4)-α-D-rhamnopyranose 29 (which is a precursor of the phosphodisaccharide 8; Scheme 4) was synthesized using acetobromogalactose 21 and methyl 2,3-O-isopropylidene-α-D-rhamnopyranoside1327 as starting materials. Their coupling in the presence of Hg(CN)2–HgBr2 in acetonitrile–toluene gave the disaccharide 28 (74%), which was converted to the crystalline heptaacetate 29 in 69% yield by acid hydrolysis followed by acetolysis/acetylation14 with 1.32% (v/v) H2SO4 in acetic anhydride.
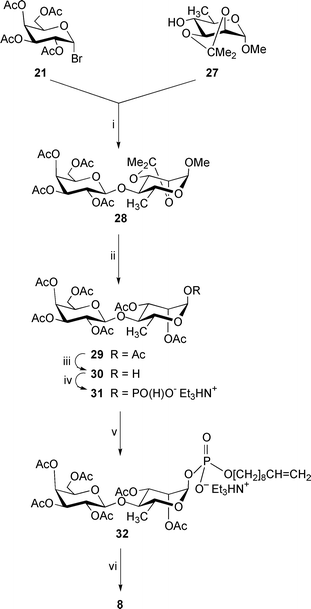 | ||
| Scheme 4 Reagents: i, Hg(CN)2, HgBr2, MeCN–PhMe; ii, (a) aq. TFA, CHCl3; (b) H2SO4, Ac2O; iii, Me2NH, MeCN–THF; iv, (a) triimidazolylphosphine, MeCN; (b) Et3NHHCO3, water (pH 7); v, (a) dec-9-en-1-ol, trimethylacetyl chloride, pyridine; (b) I2, pyridine–water; vi, NaOMe, MeOH. | ||
The hepta-O-benzoyl-β-D-fucopyranosyl-(1→4)-α-D-mannopyranose 37 (which is a precursor of the phosphodisaccharide 9; Scheme 5) was prepared in 62% yield by the glycosylation of the D-mannose tetrabenzoate336 with the α-D-fucosyl trichloroacetimidate 35 in the presence of trimethylsilyl (TMS) triflate. A small proportion of the isomeric α-linked disaccharide 40 (13%; a precursor of the phosphodisaccharide 10; Scheme 6) was also isolated from the reaction mixture. The trichloroacetimidate 35 in turn was synthesized from D-fucose by consecutive standard benzoylation ( → 33), anomeric deprotection with Me2NH ( → 34; 61%) and the reaction (93% yield) with trichloroacetonitrile in the presence of 1,8–diazabicyclo[5.4.0]undec-7-ene (DBU).15
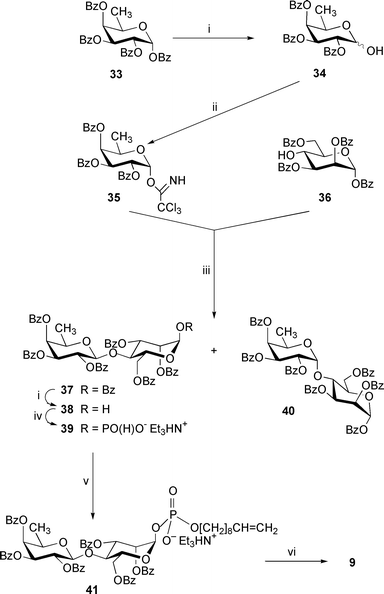 | ||
| Scheme 5 Reagents: i, Me2NH, MeCN–THF; ii, CCl3CN, DBU, CH2Cl2; iii, TMS triflate, MS 4 Å, CH2Cl2; iv, (a) triimidazolylphosphine, MeCN; (b) Et3NHHCO3, water (pH 7); v, (a) dec-9-en-1-ol, trimethylacetyl chloride, pyridine; (b) I2, pyridine–water; vi, NaOMe, MeOH. | ||
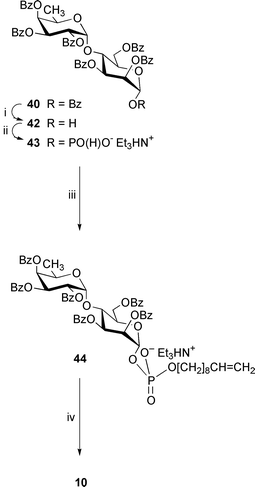 | ||
| Scheme 6 Reagents: i, Me2NH, MeCN–THF; ii, (a) triimidazolylphosphine, MeCN; (b) Et3NHHCO3, water (pH 7); iii, (a) dec-9-en-1-ol, trimethylacetyl chloride, pyridine; (b) I2, pyridine–water; iv, NaOMe, MeOH. | ||
The β-configuration of newly formed glycosidic linkages in the disaccharides 22, 28 and 37 followed from the characteristic values of J1′,2′ (7.5–8.0 Hz) in 1H NMR spectra. For the α-D-fucoside 40 the corresponding value is J1′,2′ = 3.0 Hz.
In contrast to the anomeric de-O-acylation of the peracetylated lactose 11 (see above), similar reaction of the disaccharide heptaacetate 29 and heptabenzoates 37 and 40 with dimethylamine in CH3CN–THF afforded the pure α-hemiacetal derivatives 30, 38 and 42 (66–90%), respectively. Compounds 30, 38 and 42 were then treated with triimidazolylphosphine followed by mild hydrolysis to produce the α-linked glycosyl H-phosphonates 31, 39 and 43, respectively, in 70–97% yield.
The structures of all the prepared disaccharide H-phosphonates were confirmed by NMR and mass spectrometric data (see Experimental section). For example, signals characteristic of the H-phosphonate group [δP 0.77; δH 5.67 (dd, J1,2 3.4, J1,P 8.8, 1-H), 6.89 (d, 1JH,P 637.8, HP)] were present in the 31P and 1H NMR spectra of the derivative 14. The α-configuration of the D-glucopyranosyl residue followed from the characteristic value of J1,2. The main signal in the (electrospray) ES(−) mass spectrum corresponded to the pseudomolecular ion (m/z 698.9, [M − Et3N − H]−) for the compound. The structures 23, 31, 39 and 43 were established in similar manner apart from that the α-configuration of the D-Altp (in compound 23), D-Rhap (in compound 31) and D-Manp (in compounds 39 and 43) residues followed from the characteristic positions of the 3- and 5-H resonances in 1H NMR spectra. The chemical shifts of these signals were close to those of 3- and 5-H of the disaccharide derivatives 22 (containing a benzyl 2,3,6-tri-O-benzoyl-α-D-altropyranoside moiety), 29 (containing a 1,2,3-tri-O-acetyl-α-D-rhamnopyranose moiety) and 37 and 40 (both containing 1,2,3,6-tetra-O-benzoyl-α-D-mannopyranose moieties), respectively.
The glycosyl H-phosphonates 14, 23, 31, 39 and 43 were converted to the protected phosphodiesters 15, 26, 32, 41 and 44 (75-96% yield), respectively, by their condensation with dec-9-en-1-ol in pyridine in the presence of trimethylacetyl chloride followed by oxidation of the resulting H-phosphonic diesters with iodine in aq. pyridine. The deprotected phosphodisaccharides 6–10 were prepared from the derivatives 15, 26, 32, 41 and 44, respectively, by de-O-acylation with 0.05 mol dm−3 methanolic sodium methoxide in 88–100% yield.
The structures of the compounds 6–10 and the protected phosphodiesters 15, 26, 32, 41 and 44 were confirmed by NMR and mass spectrometric data. The 31P NMR spectra exhibited single signals [δP between −1.35 and −1.96 for the deprotected compounds 6–10 (in D2O) and between −1.67 and −3.12 for the protected phosphodiesters (in CDCl3)], which are characteristic for glycoside-linked phosphodiesters.3–6,8–10 The presence of a (1→1)-phosphodiester linkage at the reducing terminus of each of the disaccharides 6–10 was confirmed by the C-1 and C-2 signals of the corresponding monosaccharide residue and the dec-9-enyl unit in the 13C NMR spectra (Table 1). These signals were shifted as a result of the α- and β-effects of phosphorylation and were coupled with phosphorus (or broadened).
| Residue | Atom |
6a |
7a |
8b |
9a |
10a |
|---|---|---|---|---|---|---|
|
a
Additional signals of Et3NH+ [δC 9.20–9.37 (CH3) and δC 47.41–47.63 (CH2)] were present.
b
Additional signals of CCH2C [δC 25.95–26.26, 29.19–30.09 and 34.16–34.44] were present.
c
Corresponds to the pseudomolecular ions [M − Et3N − H]−. For compounds 6 and 7 (triethylammonium salt), C28H56NO14P requires M, 661.34 (expected m/z, 559.14); for compounds 8–10 (triethylammonium salt), C28H56NO13P requires M, 645.35 (expected m/z, 534.15).
|
||||||
| Dec-9-enyl | OCH2CH2 |
67.20d | 67.82d | 67.76d | 67.45br | 67.01br |
| J C,P 4.0 | J C,P ≈6 | J C,P ≈6 | ||||
| OCH2CH2 |
31.15d | 30.96d | 30.87d | 30.95br | 31.03d | |
| J C,P 9.0 | J C,P 8.8 | J C,P 5.9 | J C,P 8.3 | |||
–CH![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) |
140.32 | 141.64 | 141.54 | 140.87 | 141.52 | |
| CH2 |
115.10 | 115.09 | 115.00 | 115.00 | 114.98 | |
| Aldose | C-1 | 95.71d | 96.76br | 96.70br | 96.69br | 96.78d |
| J C,P 6.9 | J C,P 5.8 | |||||
| J C,H 171.3 | J C,H 169.7 | J C,H 171.0 | J C,H 170.5 | |||
| C-2 | 72.10d | 71.54d | 71.18d | 70.89d | 71.29d | |
| J C,P 7.7 | J C,P 10.0 | J C,P 6.9 | J C,P 7.2 | J C,P 7.5 | ||
| C-3 | 72.23 | 70.95 | 69.61 | 69.76 | 70.54 | |
| C-4 | 78.77 | 74.38 | 82.62 | 77.20 | 76.98 | |
| C-5 | 72.39 | 69.78 | 69.44 | 73.12 | 73.30 | |
| C-6 | 60.65 | 61.53 | 17.82 | 61.10 | 61.30 | |
| Aldose′ | C-1′ | 103.85 | 105.04 | 104.27 | 103.87 | 102.08 |
| J C,H 162.5 | J C,H 161.0 | J C,H 160.5 | J C,H 171.0 | |||
| C-2′ | 71.83 | 71.96 | 72.09 | 71.45 | 69.64 | |
| C-3′ | 73.58 | 73.67 | 73.61 | 73.71 | 71.23 | |
| C-4′ | 69.51 | 69.96 | 69.72 | 72.12 | 72.57 | |
| C-5′ | 76.25 | 76.21 | 76.37 | 72.01 | 68.22 | |
| C-6′ | 61.93 | 62.20 | 62.18 | 16.34 | 16.42 | |
| Phosphate | P | −1.96 | −1.35 | −1.50 | −1.41 | −1.66 |
|
m/zc |
559.34 | 558.90 | 543.25 | 543.10 | 543.10 | |
The α-configuration of the D-glucopyranosyl phosphate fragments in compounds 6 and 15 was evident from the characteristic values of J1,2 = 3.4–3.5 Hz in the 1H NMR spectra (see Experimental section). The α-configuration of the D-altropyranosyl residue in the phosphodisaccharide 7 followed from the characteristic value† of 1JC,H = 171.3 Hz for the signal of C-1 and the characteristic position of the C-5 resonance of D-Altp in the 13C NMR spectrum (Table 1). The chemical shift of the C-5 signal (δC 69.78) is fairly close to that of C-5 (δC 70.00)‡ of methyl α-D-altropyranoside.17
The α-configuration of the D-mannopyranosyl phosphate fragments in compounds 9 and 10 and of the D-rhamnopyranosyl phosphate in compound 8 was confirmed by 1) the characteristic values† of 1JC,H for the signals of C-1 and 2) the characteristic positions of the C-3 and C-5 resonances of D-Manp and D-Rhap residues, respectively, in the 13C NMR spectra (see Table 1). The chemical shifts of the signals of C-3 and -5 of D-Manp and C-3 of D-Rhap (i.e., 6-deoxy-D-mannose) are close to those of C-3 and C-5 of α-D-mannopyranosyl phosphate18 taking into account the influence of the glycosyl substituents at position 4. The chemical shift of C-5 resonance (δC 69.44) of D-Rhap in compound 8 is very close to that of C-5 (δC 69.40)§
of methyl α-D-rhamnopyranoside.17
The α-configuration of the glycosyl phosphate linkages in the protected derivatives 26, 32, 41 and 44 followed from the characteristic positions of 1-, 3- and 5-H resonances in their 1H NMR spectra (see Experimental section).
The molecular masses of the phosphodiesters 6–10, 15, 26, 32, 41 and 44 were confirmed by electrospray mass spectrometry. The signals in the ES(−) mass spectra corresponded to the pseudomolecular ions for the disaccharide phosphates (see Table 1 and Experimental section). A biochemical evaluation of compounds 6–10 will be published elsewhere19 in due course.
Experimental
General procedures
Optical rotations were measured with a Perkin-Elmer 141 polarimeter; [α]D-values are given in units of 10−1 deg cm2 g−1. NMR spectra (1H at 200 and 500 MHz, 13C{1H} at 50.3 and 125 MHz, and 31P{1H} at 81 and 202.5 MHz) were recorded with Bruker AM-200 and AM-500 spectrometers for solutions in CDCl3, unless otherwise indicated. Chemical shifts (δ in ppm) are given relative to those for Me4Si (for 1H and 13C) and external aq. 85% H3PO4 (for 31P); J-values are given in Hz. ES mass spectra were recorded with a Micromass Quattro system (Micromass Biotech, UK). TLC was performed on Kieselgel 60 F254 (Merck) with A, toluene–ethyl acetate (95∶5); B, toluene–ethyl acetate (9∶1); C, toluene–ethyl acetate (7∶3); D, toluene–ethyl acetate (3∶7); E, dichloromethane–methanol (95∶5); F, chloroform–methanol (8∶2); and G, chloroform–methanol–water (10∶10∶3) as developers and detection under UV light or by charring with sulfuric acid–water–ethanol (15∶85∶5). Flash-column chromatography (FCC) was performed on Kieselgel 60 (0.040–0.063 mm) (Merck). Dichloromethane, acetonitrile and toluene were freshly distilled from CaH2. Solutions worked up were concentrated under reduced pressure at < 40 °C.2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-acetyl-α,β-D-glucopyranose 12
To a solution of 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-1,2,3,6-tetra-O-acetyl-α,β-D-glucopyranose 11 (1 g, 1.47 mmol) [prepared by standard acetylation of α-lactose with Ac2O in pyridine at 0 °C; δH (200 MHz) (inter alia) 1.94, 1.98, 2.10, 2.13 and 2.15 (15 H, 5 × s, 5 × Ac), 2.03 (9 H, s, 3 × Ac), 3.79 (t, J3,4 = J4,5 = 9.6, 4-Hα), 3.86 (1 H, dt, J5′,6′ 6.6, 5′-H), 3.93–4.17 (4 H, m, 5-H, 6-Ha and 6′-H2), 4.42 (1 H, dd, J5,6b 1.6, J6a,6b 12.4, 6-Hb), 4.45 (d, J1′,2′ 7.9, 1′-Hα), 4.54 (d, J1′,2′ 8.2, 1′-Hβ), 4.94 (1 H, dd, J3′,4′ 3.4, 3′-H), 4.98 (dd, J2,3 9.9, 2-Hα), 5.10 (dd, J2′,3′ 10.6, 2′-Hα), 5.33 (1 H, dd, J4′,5′ 0.5, 4′-H), 5.44 (dd, 3-Hα), 5.65 (d, J1,2 7.1, 1-Hβ) and 6.22 (d, J1,2 3.6, 1-Hα); α∶β ≈ 7∶1] in acetonitrile (6 cm3) was added 2 mol dm−3 Me2NH in THF (4 cm3; 7.96 mmol) and the mixture was kept at rt with monitoring by TLC (solvent D). After 4–9 h the mixture was concentrated to dryness and acetonitrile was evaporated off from the residue. FCC [ethyl acetate–toluene, (2∶8) → (8∶2)] of the residue gave the disaccharide α,β-hemiacetal12 (0.779 g, 83%) as an amorphous solid, [α]25D +35.2 (c 1.06, CHCl3) (Found: C, 48.8; H, 5.6. C26H36O18 requires C, 49.1; H, 5.7%); δH (200 MHz) (inter alia) 1.96, 2.03, 2.04, 2.05, 2.07, 2.12 and 2.15 (21 H, 7 × s, 7 × Ac), 3.75 (dd, J4,5 9.3, 4-Hα), 3.86 (1 H, dt, J5′,6′ 6.3, 5′-H), 4.00–4.22 (4 H, m, 5-H, 6-Ha and 6′-H2), 4.47 (d, J1′,2′ 7.7, 1′-Hβ), 4.48 (1 H, dd, J5,6b 3.4, J6a,6b 11.2, 6-Hb), 4.49 (d, J1′,2′ 7.9, 1′-Hα), 4.76 (m, 1- and 2-Hβ), 4.81 (dd, 2-Hα), 4.94 (1 H, dd, J3′,4′ 3.2, 3′-H), 5.09 (dd, J2′,3′ 10.6, 2′-Hβ), 5.11 (dd, J2′,3′ 10.5, 2′-Hα), 5.22 (t, J2,3 = J3,4 = 9.3, 3-Hβ), 5.34 (1 H, dd, J4′,5′ 0.5, 4′-H), 5.36 (d, J1,2 3.4, 1-Hα) and 5.51 (t, J2,3 = J3,4 = 9.7, 3-Hα); α:β = 4∶1.2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-acetyl-α-D-glucopyranosyl hydrogenphosphonate, triethylammonium salt 14
To a stirred solution of imidazole (0.65 g, 9.56 mmol) in acetonitrile (10 cm3) at 0 °C was added phosphorus trichloride (0.25 cm3, 2.87 mmol) followed by Et3N (1.4 cm3, 10.04 mmol). The mixture was stirred for 20 min, after which a solution of compound 12 (0.304 g, 0.478 mmol) in MeCN (10 cm3) was added dropwise over a period of 10–15 min at 0 °C. The mixture was stirred at rt for 30–40 min and quenched with 1 mol dm−3 triethylammonium (TEA) hydrogen carbonate (pH 7; 4 cm3). The clear solution was stirred for 15 min, CH2Cl2 (100 cm3) was added and the organic layer was washed in turn with ice-cold water (2 × 40 cm3) and cold 0.5 mol dm−3 TEA hydrogen carbonate (2 × 40 cm3), dried by filtration through cotton wool, and concentrated to give the α,β-(H-phosphonate) 13, δP 0.43 (Pα) and 1.20 (Pβ); δH (200 MHz) (inter alia) 5.20 (dd, J1,2 7.7, J1,P 9.1, 1-Hβ), 5.67 (dd, J1,2 3.4, J1,P 8.8, 1-Hα), 6.88 (d, 1JH,P 644.0, H-Pβ) and 6.91 (d, 1JH,P 637.8, H-Pα); α∶β = 4∶1.The residue was dissolved in CH3CN (15 cm3) and anhydrous H3PO3 (0.67 g, 8.17 mmol) was added. The mixture was stirred at rt for 19 h, then diluted with CH2Cl2 (100 cm3) and washed successively with cold saturated aq. NaHCO3 (2 × 40 cm3) and cold 0.5 mol dm−3 aq. TEA hydrogen carbonate (2 × 40 cm3). The organic phase (containing the hemiacetal 12) was discarded. The aqueous washings were then combined, and extracted with CH2Cl2 (4 × 40 cm3). The combined organic washings were dried by filtration through cotton wool, and concentrated to produce the α-hydrogenphosphonate 14 (0.184 g, 48%) as a chromatographically homogeneous amorphous solid, [α]26D +41.8 (c 0.97, CHCl3); δH (200 MHz) 1.32 (9 H, t, 3 × MeCH2), 1.92, 2.00, 2.02, 2.07 and 2.10 (15 H, 5 × s, 5 × Ac), 1.99 (6 H, s, 2 × Ac), 3.04 (6 H, q, 3 × MeCH2), 3.74 (1 H, t, J3,4 = J4,5 = 9.6, 4-H), 3.84 (1 H, t, J5′,6′ 6.7, 5′-H), 3.97–4.20 (4 H, m, 5-H, 6-Ha and 6′-H2), 4.41 (1 H, d, J1′,2′ 7.7, 1′-H), 4.42 (1 H, br d, J6a,6b 10.9, 6-Hb), 4.84 (1 H, dd, J1,2 3.4, 2-H), 4.90 (1 H, dd, J3′,4′ 3.3, 3′-H), 5.06 (1 H, dd, J2′,3′ 10.3, 2′-H), 5.30 (1 H, d, 4′-H), 5.44 (1 H, t, J2,3 9.6, 3-H), 5.67 (1 H, dd, J1,P 8.8, 1-H) and 6.89 (1 H, d, JH,P 637.8, HP); δP 0.77; ESMS(−) data: m/z 698.9 (100%, [M − Et3N − H]−) (expected m/z, 699.08. C32H52NO20P requires M, 801.28).
Dec-9-enyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-acetyl-α-D-glucopyranosyl phosphate, triethylammonium salt 15
A mixture of the H-phosphonate 14 (126 mg, 0.16 mmol) and dec-9-en-1-ol (0.056 cm3, 0.31 mmol) was dried by evaporation of pyridine (3 × 2 cm3) therefrom. The residue was dissolved in pyridine (1 cm3), trimethylacetyl chloride (0.048 cm3, 0.39 mmol) was added, and the mixture was stirred at rt for 10–15 min, whereafter a freshly prepared solution of iodine (80 mg, 0.314 mmol) in pyridine–water (95∶5; 2 cm3) was added. After 30 min, CH2Cl2 was added and the solution was washed successively with ice-cold 1 mol dm−3 aq. Na2S2O3 and cold 0.5 mol dm−3 aq. TEA hydrogen carbonate, dried by filtration through cotton wool, and concentrated. FCC [CH2Cl2–MeOH–Et3N, (99∶0∶1) → (89∶10∶1)] of the residue gave the phosphodiester 15 (144 mg, 96%) as an amorphous solid, [α]25D +37 (c 0.96, CHCl3); δH (200 MHz) 1.20–1.32 (10 H, m, 5 × CH2), 1.29 (9 H, t, 3 × MeCH2), 1.57 (2 H, tt, J 6.9, OCH2CH2CH2), 1.93, 2.01, 2.02, 2.09 and 2.12 (15 H, 5 × s, 5 × Ac), 2.00 (8 H, s, 2 × Ac and m, CH2CH2CH=), 3.03 (6 H, q, 3 × MeCH2), 3.76 (1 H, t, J3,4 = J4,5 = 9.6, 4-H), 3.83 (1 H, t, J5′,6′ 6.7, 5′-H), 3.85 (2 H, m, OCH2CH2), 3.98–4.12 (3 H, m, 6-Ha and 6′-H2), 4.17 (1 H, ddd, J5,6a 2.4, 5-H), 4.41 (1 H, d, J1′,2′ 7.7, 1′-H), 4.45 (1 H, dd, J5,6b 1.3, J6a,6b 12.0, 6-Hb), 4.83 (1 H, ddd, J1,2 3.4, J2,P 1.9, 2-H), 4.90 (1 H, dd, 3′-H), 4.86 (1 H, dd, 2JH,H 1.4, 3JH,H-Z 10.3, HCHCH), 4.95 (1 H, dd, 3JH,H-E 17.0, HCHCH), 5.07 (1 H, dd, J2′,3′ 10.4, 2′-H), 5.31 (1 H, d, J3′,4′ 3.2, 4′-H), 5.45 (1 H, t, J2,3 9.6, 3-H), 5.63 (1 H, dd, J1,P 8.0, 1-H) and 5.77 (1 H, ddt, JH,CH2 6.7, CH2CHCH2); δP
−1.67; ESMS(−): m/z 853.0 (100%, [M − Et3N − H]–) (expected m/z, 853.22. C42H70NO21P requires M, 955.42).
Dec-9-enyl β-D-galactopyranosyl-(1→4)-α-D-glucopyranosyl phosphate, triethylammonium salt 6
To a solution of compound 15 (138 mg) in MeOH (15 cm3) was added 0.5 mol dm−3 methanolic NaOMe (1.7 cm3). The mixture (now 0.05 mol dm−3 in NaOMe) was kept at room temperature for 1 h, whereafter it was deionized with Dowex 50W-X4 (H+) resin, filtered, and immediately neutralized with Et3N. The solution was concentrated and methanol was evaporated off from the residue. The phosphodiester 6 (96 mg, 100%) was thereby obtained as an amorphous solid, [α]25D +54.5 (c 1, MeOH); δH (200 MHz; D2O) (inter alia) 1.16 (9 H, t, 3 × MeCH2), 1.19 (10 H, m, 5 × CH2), 1.48 (2 H, tt, J 6.9, OCH2CH2CH2), 1.91 (2 H, dt, J 6.6, CH2CH2CH), 3.06 (6 H, q, 3 × MeCH2), 4.34 (1 H, d, J1′,2′ 7.6, 1′-H), 5.33 (1 H, dd, J1,2 3.5, J1,P 7.1, 1-H) and 5.71 (1 H, ddt, JH,CH2 6.6, JH,H-Z 10.2, JH,H-E 17.0, CH2CHCH2); δC, δP and ESMS(−) data: see Table 1.
Benzyl 2-O-benzoyl-4,6-O-isopropylidene-α-D-mannopyranoside 17
To a stirred solution of benzyl 4,6-O-isopropylidene-α-D-mannopyranoside 16 (3.1 g, 10 mmol) [prepared from benzyl α-D-mannopyranoside and 2-methoxypropene in 85% yield, [α]25D +85 (c 1, CHCl3), Rf 0.3 (solvent E) (Found: C, 61.8; H, 7.2. C16H22O6 requires C, 61.9; H, 7.1%) as described for the preparation of methyl 4,6-O-isopropylidene-α-D-mannopyranoside11] and BzCN (1.57 g, 12 mmol) in acetonitrile (20 cm3) was added Et3N (0.025 cm3). After 30 min, methanol was added, the reaction mixture was concentrated, and toluene was evaporated off from the residue. FCC (solvent A) gave the monobenzoate17 (3.15 g, 76%) as an amorphous solid, [α]25D +48 (c 1, CHCl3); Rf 0.2 (solvent B) (Found: C, 66.25; H, 6.3. C23H26O7 requires C, 66.65; H, 6.3%); δH (200 MHz) 1.49 and 1.61 (6 H, 2 × s, 2 × Me), 3.73-3.93 (3 H, m, 5-H and 6-H2), 4.07 (1 H, t, J3,4 = J4,5 = 9.0, 4-H), 4.26 (1 H, dd, J2,3 3.4, 3-H), 4.53 and 4.73 (2 H, AB q, J 11.7, CH2Ph), 5.00 (1 H, d, J1,2 1.3, 1-H), 5.50 (1 H, dd, 2-H) and 7.15–8.15 (10 H, m, 2 × Ph).
Benzyl 2,3-di-O-benzoyl-4,6-O-isopropylidene-α-D-altropyranoside 19
Triflic anhydride (2.05 cm3, 12.2 mmol) was added dropwise to a cooled (0 °C) stirred solution of compound 17 (2.53 g, 6.11 mmol) in CH2Cl2 (50 cm3) containing pyridine (3.85 cm3, 48.9 mmol), and then the reaction mixture was allowed to warm to rt. After 1 h, the mixture was diluted with CH2Cl2, washed successively with ice-cold 0.1 mol dm−3 HCl, ice-cold saturated aq. NaHCO3 and water, and dried by filtration through cotton wool. The filtrate was concentrated to dryness and toluene was evaporated off from the residue to produce the triflate 18 [Rf 0.5 (solvent B), δH (200 MHz) 1.45 and 1.58 (6 H, 2 × s, 2 × Me), 3.83–3.93 (3 H, m, 5-H and 6-H2), 4.28 (1 H, t, J3,4 = J4,5 = 9.1, 4-H), 4.57 and 4.72 (2 H, AB q, J 11.7, CH2Ph), 5.02 (1 H, d, J1,2 1.1, 1-H), 5.25 (1 H, dd, J2,3 3.6, 3-H), 5.65 (1 H, dd, 2-H) and 7.10–8.10 (10 H, m, 2 × Ph)].A solution of tetrabutylammonium benzoate (3.63 g, 10 mmol; dried beforehand by evaporation of anhydrous toluene therefrom) in toluene (20 cm3) was added to a solution of the triflate 18 in the same solvent (30 cm3). The reaction mixture was stirred at 60 °C for 7 h, then diluted with CH2Cl2, washed successively with saturated aq. NaHCO3 and water, dried by filtration through cotton wool and concentrated. FCC (solvent A) gave the altroside19 (2.3 g, 73%), mp 142–144 °C (from diethyl ether–hexane); [α]25D +16 (c 1, CHCl3); Rf 0.5 (solvent B) (Found: C, 69.8; H, 5.9. C30H30O8 requires C, 69.5; H, 5.8%); δH (200 MHz) 1.35 and 1.61 (6 H, 2 × s, 2 × Me), 3.90 (1 H, t, J5,6a = J6a,6b = 9.6, 6-Ha), 3.98 (1 H, dd, J5,6b 5.7, 6-Hb), 4.26 (1 H, dd, J4,5 9.6, 4-H), 4.43 (1 H, dt, 5-H), 4.53 and 4.82 (2 H, AB q, J 11.1, CH2Ph), 5.03 (1 H, d, J1,2 1.1, 1-H), 5.39 (1 H, dd, 2-H), 5.55 (1 H, t, J2,3 = J3,4 = 3.0, 3-H) and 7.15–8.15 (15 H, m, 3 × Ph).
Benzyl 2,3,6-tri-O-benzoyl-α-D-altropyranoside 20
A solution of the altroside 19 (2.49 g, 4.8 mmol) in 80% aq. acetic acid (50 cm3) was heated at 60 °C for 1 h, whereafter the mixture was concentrated and toluene was twice evaporated off from the residue. The residue was dissolved in acetonitrile (50 cm3) and BzCN (0.63 g, 4.82 mmol) and Et3N (0.025 cm3) were added to the solution. After 30 min, methanol was added, the reaction mixture was concentrated, and toluene was evaporated off from the residue. FCC (solvent A) gave the tribenzoate20 (1.87 g, 67%), mp 140–142 °C (from diethyl ether–hexane); [α]25D −6.5 (c 1, CHCl3); Rf 0.25 (solvent B) (Found: C, 70.4; H, 5.1. C34H30O9 requires C, 70.1; H, 5.2%); δH (200 MHz; CDCl3 + D2O) 4.28 (1 H, dd, J4,5 9.7, 4-H), 4.48 (1 H, ddd, J5,6a 2.3, 5-H), 4.58 and 4.83 (2 H, AB q, J 10.8, CH2Ph), 4.66 (1 H, dd, J6a,6b 12.0, 6-Ha), 4.78 (1 H, dd, J5,6b 4.0, 6-Hb), 5.10 (1 H, d, J1,2 1.0, 1-H), 5.42 (1 H, dd, 2-H), 5.62 (1 H, t, J2,3 = J3,4 = 3.2, 3-H) and 7.15–8.15 (20 H, m, 4 × Ph).Benzyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-Dl-altropyranoside 22
A solution of acetobromogalactose 21 (1.03 g, 2.5 mmol) in CH2Cl2 (15 cm3) was added dropwise to a stirred mixture of the altroside 20 (0.73 g, 1.25 mmol), Ag2CO3 (1.37 g, 5.0 mmol), AgOTf (0.64 g, 2.5 mmol) and freshly activated molecular sieves 4 Å (powder, 5 g) in boiling dichloromethane (30 cm3). The reaction mixture was stirred under reflux for 2.5 h and then at rt for 20 h. The solids were filtered off and the filtrate was concentrated. FCC [diethyl ether–hexane, (1∶1) → (2∶1)] gave the disaccharide22 (0.59 g, 52%) as an amorphous solid, [α]25D +12 (c 1, CHCl3); Rf 0.45 (solvent C) (Found: C, 63.1; H, 5.4. C48H48O18 requires C, 63.15; H, 5.3%); δH (200 MHz) 1.91 (6 H, s, 2 × Ac), 1.93 and 2.01 (6 H, 2 × s, 2 × Ac), 3.82–3.99 (3 H, m, 5′-H and 6′-H2), 4.32 (1 H, dd, J4,5 9.5, 4-H), 4.44 (1 H, dd, J5,6a 4.5, J6a,6b 12.5, 6-Ha), 4.52 and 4.81 (2 H, AB q, J 11.6, CH2Ph), 4.58–4.70 (3 H, m, 1′-, 5-H and 6-Hb), 4.91 (1 H, dd, J2′,3′ 10.5, 3′-H), 5.02 (1 H, br s, 1-H), 5.15 (1 H, dd, J1′,2′ 7.5, 2′-H), 5.25 (1 H, d, J3′,4′ 3.5, 4′-H), 5.46 (1 H, d, 2-H), 5.66 (1 H, t, J2,3 = J3,4 = 3.3, 3-H) and 7.15–8.15 (20 H, m, 4 × Ph).
2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-D-altropyranosyl hydrogenphosphonate, triethylammonium salt 23
A solution of the disaccharide 22 (0.455 g, 0.499 mmol) in THF (5 cm3) containing 20% Pd(OH)2/C (200 mg) was shaken under a slight overpressure of H2 at rt for 2 h. The catalyst was filtered off through a Celite pad and the filtrate was concentrated to give the α,β-hemiacetal 24 (0.39 g, 95%) as an amorphous solid [Rf 0.3 (solvent C); δH (500 MHz) 1.89–2.03 (12 H, m, 4 × Ac), 3.78–3.92 (3 H, m, 5′-H and 6′-H2), 4.30 (dd, J4,5 8.5, 4-Hβ), 4.39 (dd, J4,5 8.5, 4-Hα), 4.42 (dt, J5,6a = J5,6b = 3.0, 5-Hβ), 4.52 (dd, J5,6a 3.0, J6a,6b 11.7, 6-Ha), 4.61 (d, J1′,2′ 7.0, 1′-Hα), 4.67 (d, J1′,2′ 7.0, 1′-Hβ), 4.70–4.78 (m, 5-Hα and 6-Hb), 4.92 (dd, J3′,4′ 3.0, 3′-Hα), 4.95 (dd, J3′,4′ 3.0, 3′-Hβ), 5.13 (dd, J2′,3′ 9.0, 2′-Hα), 5.17 (dd, J2′,3′ 9.0, 2′-Hβ), 5.22 (d, 4′-Hα), 5.26 (d, 4′-Hβ), 5.30 (br s, 1-Hβ), 5.39 (br s, 1-Hα), 5.46 (d, 2-Hβ), 5.51 (d, 2-Hα), 5.69 (t, J2,3 = J3,4 = 2.8, 3-Hβ), 5.79 (t, J2,3 = J3,4 = 2.8, 3-Hα) and 7.20–8.20 (15 H, m, 3 × Ph); α∶β = 0.8∶1].
The reaction of the compound 24 (0.39 g, 0.474 mmol) with PCl3 (0.165 cm3, 1.89 mmol), imidazole (0.45 g, 6.62 mmol) and Et3N (0.99 cm3, 7.09 mmol) in CH3CN (10 cm3), followed by hydrolysis with 1 mol dm−3 aq. TEA hydrogen carbonate (2.5 cm3), was accomplished as described for the preparation of the disaccharide H-phosphonate 13. After work-up, the solution was concentrated and acetonitrile was evaporated off from the residue. The residue was dissolved in the same solvent (5 cm3) and anhydrous H3PO3 (0.39 g, 4.73 mmol) was added to the solution. The reaction mixture was kept at rt for 20 h, then diluted with CH2Cl2 (50 cm3) and washed successively with saturated aq. NaHCO3 and 0.5 mol dm−3 aq. TEA hydrogen carbonate, dried by filtration through cotton wool, and concentrated. FCC [CH2Cl2–MeOH, (99∶1) → (80∶20)] gave the H-phosphonate 23 (0.175 g, 35% from the disaccharide 22) as an amorphous solid, [α]25D +1 (c 1, CHCl3); Rf 0.35 (solvent F); δH (200 MHz) 1.20 (9 H, t, 3 × MeCH2), 1.91, 1.92, 1.93 and 2.02 (12 H, 4 × s, 4 × Ac), 2.91 (6 H, q, 3 × MeCH2), 3.80–3.88 (2 H, m, 5′-H and 6′-Ha), 3.93 (1 H, dd, J5′,6b′ 7.8, J6a′,6b′ 13.5, 6′-Hb), 4.35 (1 H, dd, J4,5 9.6, 4-H), 4.46 (1 H, dd, J6a,6b 11.6, 6-Ha), 4.64 (1 H, d, J1′,2′ 7.7, 1′-H), 4.71 (1 H, dd, J5,6b 1.0, 6-Hb), 4.88 (1 H, ddd, J5,6a 3.7, 5-H), 4.91 (1 H, dd, J3′,4′ 3.2, 3′-H), 5.13 (1 H, dd, J2′,3′ 10.6, 2′-H), 5.23 (1 H, d, 4′-H), 5.44 (1 H, d, 2-H), 5.67 (1 H, t, J2,3 = J3,4 = 2.8, 3-H), 5.72 (1 H, d, J1,P 8.5, 1-H), 7.02 (1 H, d, JH,P 640.0, HP) and 7.40–8.20 (15 H, m, 3 × Ph); δP 0.58; ESMS(−): m/z 884.9 (100%, [M − Et3N − H]−) (expected m/z, 885.008. C47H58NO20P requires M, 987.208). Also isolated was the disaccharide hemiacetal 24 (0.2 g, 49% recovery).
Dec-9-enyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-D-altropyranosyl phosphate, triethylammonium salt 26
This compound was prepared by condensation of the glycobiosyl H-phosphonate 23 (98 mg, 0.10 mmol) and dec-9-en-1-ol (0.053 cm3, 0.30 mmol) in pyridine (1 cm3) in the presence of trimethylacetyl chloride (0.037 cm3, 0.30 mmol), followed by oxidation with iodine (51 mg, 0.20 mmol) in pyridine–water (95∶5; 2 cm3) as described for the synthesis of the phosphodiester 15. FCC [CH2Cl2–MeOH, (99∶1) → (80∶20)] gave the phosphodiester 26 (85 mg, 75%) as an amorphous solid, [α]25D +2 (c 1, CHCl3); Rf 0.5 (solvent F); δH (200 MHz) 1.25 (19 H, m, 3 × MeCH2 and 5 × CH2), 1.48 (2 H, tt, J 6.9, OCH2CH2CH2), 1.95 (6 H, s, 2 × Ac), 1.97 and 2.00 (6 H, 2 × s, 2 × Ac), 2.03 (2 H, m, CH2CH2CH), 2.95 (6 H, q, 3 × MeCH2), 3.74–3.96 (5 H, m, 5′-H, 6′-H2 and OCH2CH2), 4.38 (1 H, dd, J4,5 9.6, 4-H), 4.46 (1 H, dd, J5,6a 3.0, J6a,6b 11.8, 6-Ha), 4.64 (1 H, d, J1′,2′ 7.8, 1′-H), 4.72 (1 H, dd, J5,6b 1.0, 6-Hb), 4.87–4.95 (3 H, m, 3′-, 5-H and HCHCH), 4.98 (1 H, dd, 2JH,H 1.6, 3JH,H-E 16.8, HCHCH), 5.12 (1 H, dd, J2′,3′ 10.1, 2′-H), 5.23 (1 H, d, J3′,4′ 3.1, 4′-H), 5.50 (1 H, d, J2,3 3.0, 2-H), 5.64 (1 H, d, J1,P 7.7, 1-H), 5.68 (1 H, dd, J3,4 3.5, 3-H), 5.80 (1 H, ddt, JH,CH2 6.6, 3JH,H-Z 10.9, CH2CHCH2) and 7.40–8.25 (15 H, m, 3 × Ph); δP
−2.79; ESMS(−): m/z 1039.0 (100%, [M − Et3N − H]−) (expected m/z, 1039.14. C57H76NO21P requires M, 1141.344).
Dec-9-enyl β-D-galactopyranosyl-(1→4)-α-D-altropyranosyl phosphate, triethylammonium salt 7
To a solution of compound 26 (50 mg) in MeOH (1.8 cm3) was added 0.5 mol dm−3 methanolic NaOMe (0.2 cm3). The mixture (now 0.05 mol dm−3 in NaOMe) was kept at 0 °C for 16 h and then at room temperature for 8 h, whereafter it was deionized with Dowex 50W-X4(H+) resin, filtered and immediately neutralized with Et3N. After concentration, water (3 × 5 cm3) was evaporated off from the residue to remove methyl benzoate. The phosphodiester 7 (28 mg, 96%) was thereby obtained as an amorphous solid, [α]25D +36 (c 1, MeOH); Rf 0.65 (solvent G); δH (200 MHz; D2O) (inter alia) 1.15 (9 H, t, 3 × MeCH2), 1.23 (10 H, m, 5 × CH2), 1.52 (2 H, tt, J 6.9, OCH2CH2CH2), 1.94 (2 H, dt, J 6.7, CH2CH2CH), 3.10 (6 H, q, 3 × MeCH2), 4.41 (1 H, d, J1′,2′ 7.0, 1′-H), 5.21 (1 H, br d, J1,P 6.6, 1-H) and 5.83 (1 H, ddt, JH,CH2 6.7, JH,H-Z 10.1, JH,H-E 18.0, CH2CHCH2); δC, δP and ESMS(−) data: see Table 1.
Methyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3-O-isopropylidene-α-D-rhamnopyranoside 28
A solution of acetobromogalactose 21 (1.49 g, 3.58 mmol) in acetonitrile–toluene (10∶3; 13 cm3) was added to a stirred mixture of methyl 2,3-O-isopropylidene-α-D-rhamnopyranoside1327 (0.318 g, 1.45 mmol), Hg(CN)2 (0.9 g, 3.58 mmol) and HgBr2 (0.64 g, 1.79 mmol) in the same mixed solvent (5 cm3). After being stirred at rt for 16 h, the reaction mixture was diluted with CH2Cl2 (50 cm3), washed successively with 1 mol dm−3 aq. KBr, saturated aq. NaHCO3, and water, dried by filtration through cotton wool, and concentrated. FCC [toluene–ethyl acetate, (8∶2)] of the residue gave the disaccharide derivative28 (0.59 g, 74%), mp 126–129 °C (from ethanol); [α]22D +25.7 (c 1, CHCl3) (Found: C, 52.5; H, 6.6. C24H36O14 requires C, 52.6; H, 6.6%); δH (200 MHz) 1.23 (3 H, d, J5,6 6.3, 6-H3), 1.32 and 1.50 (6 H, 2 × s, CMe2), 2.00, 2.05, 2.07 and 2.15 (12 H, 4 × s, 4 × Ac), 3.34 (3 H, s, OMe), 3.36 (1 H, dd, J3,4 7.2, 4-H), 3.63 (1 H, dq, J4,5 9.8, 5-H), 3.88 (1 H, t, J5′,6′ 6.6, 5′-H), 4.06 (1 H, d, J2,3 5.7, 2-H), 4.14 (2 H, d, 6′-H2), 4.24 (1 H, dd, 3-H), 4.65 (1 H, d, J1′,2′ 8.0, 1′-H), 4.81 (1 H, s, 1-H), 5.00 (1 H, dd, J3′,4′ 3.2, 3′-H), 5.21 (1 H, dd, J2′,3′ 10.3, 2′-H) and 5.36 (1 H, d, 4′-H).
2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-1,2,3-tri-O-acetyl-α-D-rhamnopyranose 29
To a stirred solution of the disaccharide 28 (0.728 g) in chloroform (36 cm3) was added 90% aq. trifluoroacetic acid (4 cm3). Stirring was continued for 2 h, whereafter the solution was concentrated and toluene was twice evaporated off from the residue. The residue was then dissolved in Ac2O (5 cm3) and a sulfuric acid–acetic anhydride mixture [1∶50 (v/v); 10.2 cm3] was added. The solution was stirred at rt for 2 h, whereafter it was diluted with CH2Cl2 (100 cm3), washed successively with water, saturated aq. NaHCO3, and water, dried by filtration through cotton wool, and concentrated. Toluene was twice evaporated off from the residue. FCC (solvent B → solvent C) of the residue gave the heptaacetate29 (0.57 g, 69%), mp 145–148 °C (from ethanol); [α]21D +36.8 (c 0.99, CHCl3) (Found: C, 50.6; H, 5.9. C26H36O17 requires C, 50.3; H, 5.9%); δH (200 MHz) 1.30 (3 H, d, J5,6 6.1, 6-H3), 1.96, 2.02, 2.03, 2.04 and 2.14 (15 H, 5 × s, 5 × Ac), 2.13 (6 H, s, 2 × Ac), 3.63 (1 H, t, J3,4 = J4,5 = 9.4, 4-H), 3.82 (1 H, dq, 5-H), 3.87 (1 H, ddd, J5′,6a′ 7.1, 5′-H), 4.02 (1 H, dd, J6a′,6b′ 11.0, 6′-Ha), 4.16 (1 H, dd, J5′,6b′ 6.4, 6′-Hb), 4.58 (1 H, d, J1′,2′ 7.8, 1′-H), 4.97 (1 H, dd, J3′,4′ 3.4, 3′-H), 5.14 (1 H, dd, J2′,3′ 10.3, 2′-H), 5.19 (1 H, dd, J2,3 3.3, 2-H), 5.28 (1 H, dd, 3-H), 5.33 (1 H, dd, J4′,5′ 0.5, 4′-H) and 5.94 (1 H, d, J1,2 1.9, 1-H).
2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3-di-O-acetyl-α-D-rhamnopyranose 30
This compound was prepared from compound 29 (0.307 g) as described for the hemiacetal derivative 12. Two consecutive FCC separations [toluene–ethyl acetate, (55∶45) → (8∶2) and solvent B → solvent D] gave the disaccharide α-hemiacetal30 (0.19 g, 66%) as an amorphous solid, [α]21D +11 (c 0.97, CHCl3) (Found: C, 49.4; H, 6.0. C24H34O16 requires C, 49.8; H, 5.9%); δH (200 MHz) 1.28 (3 H, d, J5,6 6.2, 6-H3), 1.96, 2.01, 2.03, 2.04, 2.12 and 2.14 (18 H, 6 × s, 6 × Ac), 3.59 (1 H, t, J3,4 = J4,5 = 9.4, 4-H), 3.87 (1 H, t, J5′,6a′ = J5′,6b′ = 6.5, 5′-H), 4.00 (1 H, dq, 5-H), 4.01 (1 H, dd, 6′-Ha), 4.16 (1 H, dd, J6a′,6b′ 11.0, 6′-Hb), 4.57 (1 H, d, J1′,2′ 7.8, 1′-H), 4.97 (1 H, dd, J3′,4′ 3.3, 3′-H), 5.08 (1 H, d, J1,2 1.9, 1-H), 5.13 (1 H, dd, J2′,3′ 10.5, 2′-H), 5.21 (1 H, dd, J2,3 3.5, 2-H), 5.32 (1 H, d, 4′-H) and 5.33 (1 H, dd, 3-H).2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3-di-O-acetyl-α-D-rhamnopyranosyl hydrogenphosphonate, triethylammonium salt 31
This compound was prepared by the reaction of the hemiacetal 30 (0.184 g, 0.32 mmol) with PCl3 (0.166 cm3, 1.91 mmol), imidazole (0.433 g, 6.36 mmol) and Et3N (0.93 cm3, 6.7 mmol) in acetonitrile (15 cm3) followed by hydrolysis as described for the H-phosphonate 13. For work-up, the reaction mixture was diluted with CH2Cl2 (100 cm3) and washed successively with cold saturated aq. NaHCO3 (2 × 40 cm3) and cold 0.5 mol dm−3 aq. TEA hydrogen carbonate (2 × 40 cm3). The organic phase was discarded. The aqueous washings were combined, and extracted with CH2Cl2 (4 × 20 cm3). The combined organic washings were dried by filtration through cotton wool, and concentrated to produce the H-phosphonate 31 (0.165 g, 70%) as a chromatographically homogeneous amorphous solid, [α]25D +23.1 (c 1.06, CHCl3); δH (200 MHz) 1.27 (3 H, d, J5,6 6.3, 6-H3), 1.30 (9 H, t, 3 × MeCH2), 1.94, 1.95, 2.00, 2.02, 2.08 and 2.11 (18 H, 6 × s, 6 × Ac), 3.03 (6 H, q, 3 × MeCH2), 3.56 (1 H, t, J3,4 = J4,5 = 9.4, 4-H), 3.83 (1 H, t, J5′,6a′ = J5′,6b′ = 6.6, 5′-H), 3.98 (1 H, dq, 5-H), 3.99 (1 H, dd, 6′-Ha), 4.12 (1 H, dd, J6a′,6b′ 11.0, 6′-Hb), 4.54 (1 H, d, J1′,2′ 7.7, 1′-H), 4.94 (1 H, dd, J3′,4′ 3.3, 3′-H), 5.10 (1 H, dd, J2′,3′ 10.5, 2′-H), 5.21 (1 H, dd, J1,2 1.8, 2-H), 5.30 (1 H, d, 4′-H), 5.31 (1 H, dd, J2,3 3.6, 3-H), 5.44 (1 H, dd, J1,P 8.8, 1-H) and 5.44 (1 H, d, JH,P 642.0, HP); δP −0.10; ESMS(−): m/z 641.0 (100%, [M − Et3N − H]−) (expected m/z, 641.07. C30H50NO18P requires M, 743.27).Dec-9-enyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3-di-O-acetyl-α-D-rhamnopyranosyl phosphate, triethylammonium salt 32
This compound was prepared by condensation of the H-phosphonate 31 (0.14 g, 0.19 mmol) and dec-9-en-1-ol (0.067 cm3, 0.38 mmol) in pyridine (1 cm3) in the presence of trimethylacetyl chloride (0.058 cm3, 0.47 mmol) followed by oxidation with iodine (0.096 g, 0.376 mmol) in pyridine–water (95∶5; 2 cm3) as described for the synthesis of the phosphodiester 15. FCC [CH2Cl2–MeOH–Et3N, (99∶0∶1) → (89∶10∶1)] gave the phosphodiester 32 (0.148 g, 88%) as an amorphous solid, [α]27D +12.4 (c 1.09, CHCl3); δH (200 MHz) 1.18 (3 H, d, J5,6 6.0, 6-H3), 1.24 (9 H, t, 3 × MeCH2), 1.25 (10 H, m, 5 × CH2), 1.53 (2 H, tt, J 6.9, OCH2CH2CH2), 1.89, 1.90, 1.97, 1.98, 2.03 and 2.07 (18 H, 6 × s, 6 × Ac), 1.96 (2 H, dt, J 6.8, CH2CH2CH), 3.00 (6 H, q, 3 × MeCH2), 3.51 (1 H, dd, J4,5 9.7, 4-H), 3.71–3.86 (4 H, m, 5′-H, 6′-Ha and OCH2CH2), 3.97 (1 H, dq, 5-H), 4.08 (1 H, dd, J5′,6b′ 6.6, J6a′,6b′ 11.2, 6′-Hb), 4.52 (1 H, d, J1′,2′ 7.7, 1′-H), 4.84 (1 H, dd, 2JH,H 1.6, 3JH,H-Z 9.3, HCHCH), 4.90 (1 H, dd, 3JH,H-E 17.0, HCHCH), 4.92 (1 H, dd, 3′-H), 5.08 (1 H, dd, J2′,3′ 10.5, 2′-H), 5.19 (1 H, dd, J2,3 3.3, 2-H), 5.25 (1 H, d, J3′,4′ 4.0, 4′-H), 5.26 (1 H, dd, J3,4 9.4, 3-H), 5.34 (1 H, dd, J1,2 1.5, J1,P 8.8, 1-H) and 5.73 (1 H, ddt, JH,CH2 6.8, CH2CHCH2); δP
−3.12; ESMS(−): m/z 795.3 (100%, [M − Et3N − H]−) (expected m/z, 795.21. C40H68NO19P requires M, 897.41).
Dec-9-enyl β-D-galactopyranosyl-(1→4)-α-D-rhamnopyranosyl phosphate, ammonium salt 8
De-O-acetylation of compound 32 (74 mg) with 0.05 mol dm−3 NaOMe in methanol (3 h at rt) followed by work-up, as described in the preparation of the phosphodiester 6, produced a crude product, which then was applied to a column (18 × 1.5 cm) of Fractogel TSK DEAE-650 (S) (HCO3−-form) (Merck) eluted with a linear gradient of NH4HCO3 (0 → 0.1 mol dm−3) in 3∶2 water–propan-2-ol at 1 cm3 min−1 to afford the phosphodiester 8 (41 mg, 88%) as an amorphous solid, [α]26D +22.4 (c 0.99, MeOH); δH (200 MHz; D2O) (inter alia) 1.22–1.45 (13 H, m, 6-H3 and 5 × CH2), 1.63 (2 H, tt, J 6.9, OCH2CH2CH2), 2.05 (2 H, dt, J 6.9, CH2CH2CH), 4.48 (1 H, d, J1′,2′ 7.0, 1′-H), 5.32 (1 H, br d, J1,P 6.3, 1-H) and 5.91 (1 H, m, CH2CHCH2); δC, δP and ESMS(−) data: see Table 1.
2,3,4-Tri-O-benzoyl-α,β-D-fucopyranose 34
To a solution of 1,2,3,4-tetra-O-benzoyl-α-D-fucopyranose 33 (0.5 g, 0.861 mmol) [prepared by standard benzoylation of α-D-fucose with benzoyl chloride in pyridine–chloroform; δH (200 MHz) 1.34 (3 H, d, J5,6 6.4, 6-H3), 4.67 (1 H, q, 5-H), 5.92 (1 H, d, 4-H), 6.01 (1 H, dd, J2,3 10.8, 2-H), 6.14 (1 H, dd, J3,4 3.0, 3-H), 6.91 (1 H, d, J1,2 3.3, 1-H) and 7.17–8.28 (20 H, m, 4 × Ph)] in acetonitrile (3 cm3) was added 2 mol dm−3 Me2NH in THF (4.3 cm3; 8.61 mmol) and the mixture was kept at rt with monitoring by TLC (solvents B and C). After 16–24 h, the mixture was concentrated to dryness and acetonitrile was evaporated off from the residue. FCC [toluene–ethyl acetate, (99∶1) → (85∶15)] gave the unchanged starting material 33 (0.079 g, 16% recovery) and the hemiacetal34 (0.25 g, 61%; amorphous solid), [α]22D +247.4 (c 1, CHCl3) (Found: C, 68.3; H, 5.1. C27H24O8 requires C, 68.1; H, 5.1%); δH (200 MHz; CDCl3 + D2O) 1.26 (d, J5,6 6.4, 6-Hα), 1.35 (d, J5,6 6.3, 6-Hβ), 4.12 (dq, J4,5 0.8, 5-Hβ), 4.67 (q, H-5α), 5.06 (d, J1,2 6.8, 1-Hβ), 5.66–5.85 (m, 1-Hα, 2-H, 3-Hβ and 4-H), 6.09 (dd, J2,3 10.6, J3,4 3.2, 3-Hα) and 7.01–8.40 (15 H, m, 3 × Ph); α∶β = 5∶1.
2,3,4-Tri-O-benzoyl-α-D-fucopyranosyl trichloroacetimidate 35
To a stirred solution of the hemiacetal 34 (0.228 g, 0.48 mmol) and CCl3CN (2 cm3, 20 mmol) in dichloromethane (4 cm3) cooled to 0 °C was added DBU (0.072 cm3, 0.48 mmol) under argon. The mixture was stirred for 2 h at 0 °C and then concentrated. FCC (solvent A) of the residue gave the α-fucopyranosyl trichloroacetimidate 35 (0.277 g, 93%) as an amorphous solid, [α]22D +209.4 (c 1, CHCl3); δH (200 MHz) 1.34 (3 H, d, J5,6 6.5, 6-H3), 4.66 (1 H, dq, 5-H), 5.89 (1 H, dd, J4,5 0.5, 4-H), 5.92 (1 H, dd, J2,3 10.5, 2-H), 6.06 (1 H, dd, J3,4 3.2, 3-H), 6.86 (1 H, d, J1,2 3.3, 1-H), 7.10–8.23 (15 H, m, 3 × Ph) and 8.60 (1 H, s, HN); δC 16.19 (C-6), 67.95 (C-5), 68.05 (C-2), 68.66 (C-3), 71.29 (C-4), 90.91 (CCl3), 94.11 (C-1), 128.30–133.48 (Ph), 160.78 (C=NH) and 165.62–165.83 (C=O); ESMS(+): m/z 459.2 (100%, [M − CCl3CONH]+) (expected m/z, 459.47. C29H24Cl3NO8 requires M, 620.86).2,3,4-Tri-O-benzoyl-β-D-fucopyranosyl-(1→4)-1,2,3,6-tetra-O-benzoyl-α-D-mannopyranose 37 and 2,3,4-tri-O-benzoyl-α-D-fucopyranosyl-(1→4)-1,2,3,6-tetra-O-benzoyl-α-D-mannopyranose 40
A mixture of the trichloroacetimidate 35 (0.554 g, 0.89 mmol), 1,2,3,6-tetra-O-benzoyl-α-D-mannopyranose336 (0.638 g, 1.07 mmol) and freshly activated molecular sieves 4 Å (powder, 1 g) in dry dichloromethane (5 cm3) was stirred under argon for 30 min. TMS triflate (0.046 cm3, 0.22 mmol) was then added and the mixture was cooled to −70 °C. Stirring was continued for a further 1.5 h, while the mixture slowly warmed to −10 °C. The reaction was quenched with a few drops of N,N-diisopropylethylamine, the solids were filtered off, and the solvent was removed under reduced pressure. FCC (toluene → solvent B) of the residue gave a mixture of the disaccharides 37 and 40, which were then separated by further FCC [dichloromethane–ethyl acetate, (100∶0) → (98∶2)]. That provided, first, the β-linked disaccharide37 (0.582 g, 62%) as an amorphous solid, [α]23D +118 (c 1.03, CHCl3) (Found: C, 69.6; H, 4.7. C61H50O17 requires C, 69.4; H, 4.8%); δH (200 MHz) 0.82 (3 H, d, J5′,6′ 6.3, 6′-H3), 3.56 (1 H, q, 5′-H), 4.27 (1 H, ddd, J5,6a 2.6, 5-H), 4.45 (1 H, dd, J6a,6b 12.7, 6-Ha), 4.68 (1 H, dd, J5,6b 1.8, 6-Hb), 4.69 (1 H, J3,4 = J4,5 = 9.7, 4-H), 4.96 (1 H, d, J1′,2′ 7.9, 1′-H), 5.39 (1 H, dd, J3′,4′ 3.3, 3′-H), 5.48 (1 H, d, 4′-H), 5.70 (1 H, dd, J2′,3′ 10.3, 2′-H), 5.85 (1 H, dd, J2,3 3.3, 2-H), 5.99 (1 H, dd, 3-H), 6.48 (1 H, d, J1,2 1.7, 1-H) and 7.08–8.22 (35 H, m, 7 × Ph). Continued elution gave the α-linked disaccharide40 (0.126 g, 13%) as an amorphous solid, [α]23D +124 (c 0.93, CHCl3) (Found: C, 69.9; H, 5.1%); δH (200 MHz) 1.13 (3 H, d, J5′,6′ 6.6, 6′-H3), 4.42–4.56 (2 H, m, 5- and 5′-H), 4.66 (1 H, dd, J5,6a 2.7, J6a,6b 12.3, 6-Ha), 4.91 (1 H, J3,4 = J4,5 = 9.3, 4-H), 4.93 (1 H, dd, J5,6b 0.5, 6-Hb), 5.67–5.94 (6 H, 1′-, 2-, 2′-, 3-, 3′- and 4′-H), 6.55 (1 H, d, J1,2 2.0, 1-H) and 7.05–8.28 (35 H, m, 7 × Ph).
2,3,4-Tri-O-benzoyl-β-D-fucopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-D-mannopyranose 38
This compound was prepared from compound 37 (0.307 g) as described for the hemiacetal derivative 34. FCC [toluene → solvent C] gave the disaccharide α-hemiacetal38 (0.25 g, 90%) as an amorphous solid, [α]25D +123.3 (c 1.06, CHCl3) (Found: C, 68.1; H, 5.0. C54H46O16 requires C, 68.2; H, 4.9%); δH (200 MHz) 0.83 (3 H, d, J5′,6′ 6.2, 6′-H3), 3.53 (1 H, q, 5′-H), 4.07 (1 H, d, J1,OH 4.1, 1-OH), 4.33–4.46 (2 H, m, 5-H and 6-Ha), 4.58 (1 H, J3,4 = J4,5 = 9.6, 4-H), 4.77 (1 H, dd, J5,6b 0.6, J6a,6b 12.5, 6-Hb), 4.95 (1 H, d, J1′,2′ 7.9, 1′-H), 5.35 (1 H, d, J1,2 1.8, 1-H), 5.42 (1 H, dd, J2′,3′ 10.2, 3′-H), 5.46 (1 H, d, J3′,4′ 3.2, 4′-H), 5.62–5.75 (2 H, m, 2- and 2′-H), 5.95 (1 H, dd, J2,3 3.0, 3-H) and 6.98–8.22 (30 H, m, 6 × Ph).
2,3,4-Tri-O-benzoyl-β-D-fucopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-D-mannopyranosyl hydrogenphosphonate, triethylammonium salt 39
This compound was prepared from the hemiacetal 38 (0.208 g, 0.219 mmol) as described for the H-phosphonate derivative 13. This produced the disaccharide hydrogenphosphonate 39 (0.232 g, 95%) as a chromatographically homogeneous amorphous solid, [α]25D +102.4 (c 0.96, CHCl3); δH (200 MHz) 0.81 (3 H, d, J5′,6′ 6.3, 6′-H3), 1.27 (9 H, t, 3 × MeCH2), 3.01 (6 H, q, 3 × MeCH2), 3.49 (1 H, q, 5′-H), 4.37 (1 H, ddd, J5,6a 2.7, 5-H), 4.43 (1 H, dd, J6a,6b 12.9, 6-Ha), 4.53 (1 H, J3,4 = J4,5 = 9.5, 4-H), 4.65 (1 H, dd, J5,6b 1.4, 6-Hb), 4.87 (1 H, d, J1′,2′ 7.9, 1′-H), 5.34 (1 H, dd, J3′,4′ 3.4, 3′-H), 5.42 (1 H, d, 4′-H), 5.64 (1 H, dd, J2′,3′ 10.4, 2′-H), 5.67 (1 H, dd, J1,2 2.0, 2-H), 5.70 (1 H, dd, J1,P 7.7, 1-H), 5.88 (1 H, dd, J2,3 3.3, 3-H), 7.01 (1 H, d, JH,P 636.9, HP) and 7.05–8.15 (30 H, m, 6 × Ph); δP 0.11; ESMS(−): m/z 1012.8 (100%, [M − Et3N − H]−) (expected m/z, 1013.17. C60H62NO18P requires M, 1115.37).Dec-9-enyl 2,3,4-tri-O-benzoyl-β-D-fucopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-D-mannopyranosyl phosphate, triethylammonium salt 41
This compound was prepared by condensation of the H-phosphonate 39 (0.12 g, 0.107 mmol) and dec-9-en-1-ol (0.038 cm3, 0.215 mmol) in pyridine (1 cm3) in the presence of trimethylacetyl chloride (0.033 cm3, 0.268 mmol) followed by oxidation with iodine (0.055 g, 0.215 mmol) in pyridine–water (95∶5; 2 cm3) as described for the synthesis of the phosphodiester 15. FCC [CH2Cl2–MeOH–Et3N, (99∶0∶1) → (89∶10∶1)] gave the phosphodiester 41 (0.108 g, 80%) as an amorphous solid, [α]25D +82.2 (c 1, CHCl3); δH (200 MHz) 0.84 (3 H, d, J5′,6′ 6.3, 6′-H3), 1.24 (10 H, m, 5 × CH2), 1.30 (9 H, t, 3 × MeCH2), 1.58 (2 H, tt, J 6.9, OCH2CH2), 1.97 (2 H, dt, J 6.9, CH2CH2CH), 3.05 (6 H, q, 3 × MeCH2), 3.47 (1 H, q, 5′-H), 3.90 (2 H, m, OCH2CH2), 4.37–4.49 (2 H, m, 5-H and 6-Ha), 4.55 (1 H, J3,4 = J4,5 = 9.4, 4-H), 4.64 (1 H, dd, J5,6b 1.1, J6a,6b 11.7, 6-Hb), 4.87 (1 H, d, J1′,2′ 7.8, 1′-H), 4.89 (1 H, dd, 2JH,H 1.3, 3JH,H-Z 10.4, HCHCH), 4.95 (1 H, dd, 3JH,H-E 17.2, HCHCH), 5.33 (1 H, dd, J2′,3′ 10.4, 3′-H), 5.43 (1 H, d, J3′,4′ 3.3, 4′-H), 5.65 (1 H, dd, J1,P 8.3, 1-H), 5.66 (1 H, dd, 2′-H), 5.74 (1 H, dd, J1,2 2.5, 2-H), 5.78 (1 H, ddt, JH,CH2 6.9, CH2CHCH2), 5.91 (1 H, dd, J2,3 3.4, 3-H) and 7.05–8.10 (30 H, m, 6 × Ph); δP
−2.83; ESMS(−): m/z 1166.9 (100%, [M − Et3N − H]−) (expected m/z, 1167.31. C70H80NO19P requires M, 1269.51).
Dec-9-enyl β-D-fucopyranosyl-(1→4)-α-D-mannopyranosyl phosphate, triethylammonium salt 9
De-O-benzoylation of compound 41 (101 mg) with 0.05 mol dm−3 NaOMe in methanol (16 h at rt) followed by work-up, as described in the preparation of the phosphodiester 7, gave the phosphodiester 9 (51 mg, 99%) as an amorphous solid, [α]28D +22.5 (c 0.99, MeOH); δH (D2O) (inter alia) 1.22–1.39 (22 H, m, 6′-H3, 3 × MeCH2 and 5 × CH2), 1.58 (2 H, tt, J 6.9, OCH2CH2CH2), 2.01 (2 H, dt, J 6.9, CH2CH2CH), 3.15 (6 H, q, 3 × MeCH2), 4.38 (1 H, d, J1′,2′ 7.6, 1′-H), 5.37 (1 H, br d, J1,P 7.1, 1-H) and 5.83 (1 H, m, CH2CHCH2); δC, δP and ESMS(−) data: see Table 1.
2,3,4-Tri-O-benzoyl-α-D-fucopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-D-mannopyranose 42
This compound was prepared from compound 40 (160 mg) as described for the hemiacetal derivative 34. FCC [toluene → solvent C] gave the disaccharide α-hemiacetal40 (99 mg, 69%) as an amorphous solid, [α]26D +54.7 (c 1.02, CHCl3) (Found: C, 68.0; H, 5.0. C54H46O16 requires C, 68.2; H, 4.9%); δH (200 MHz) 1.13 (3 H, d, J5′,6′ 6.4, 6′-H3), 3.83 (1 H, d, J1,OH 4.3, 1-OH), 4.52 (1 H, q, 5′-H), 4.60 (1 H, ddd, J5,6b 0.7, 5-H), 4.65 (1 H, dd, J5,6a 2.6, 6-Ha), 4.81 (1 H, J3,4 = J4,5 = 9.6, 4-H), 5.01 (1 H, dd, J6a,6b 11.2, 6-Hb), 5.40 (1 H, d, J1,2 1.8, 1-H), 5.68 (1 H, dd, 2-H), 5.75 (1 H, dd, J2,3 3.4, 3-H), 5.76–5.85 (4 H, m, 1′-, 2′-, 3′- and 4′-H) and 7.10–8.25 (30 H, m, 6 × Ph).
2,3,4-Tri-O-benzoyl-α-D-fucopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-D-mannopyranosyl hydrogenphosphonate, triethylammonium salt 43
This compound was prepared from the hemiacetal 42 (88 mg, 0.092 mmol) as described for the H-phosphonate derivative 13. This produced the disaccharide hydrogenphosphonate 43 (100 mg, 97%) as a chromatographically homogeneous amorphous solid, [α]21D +61 (c 1.06, CHCl3); δH (200 MHz) 1.08 (3 H, d, J5′,6′ 6.3, 6′-H3), 1.29 (9 H, t, 3 × MeCH2), 3.01 (6 H, q, 3 × MeCH2), 4.47 (1 H, q, 5′-H), 4.57–4.68 (2 H, m, 5-H and 6-Ha), 4.79 (1 H, J3,4 = J4,5 = 9.4, 4-H), 4.93 (1 H, dd, J5,6b 2.8, J6a,6b 12.9, 6-Hb), 5.67 (1 H, dd, J1,2 2.0, J2,3 3.0, 2-H), 5.70–5.82 (6 H, m, 1-, 1′-, 2′-, 3-, 3′- and 4′-H), 7.10 (1 H, d, JH,P 640.4, HP) and 6.92–8.23 (30 H, m, 6 × Ph); δP 0.57; ESMS(−): m/z 1013.0 (100%, [M − Et3N − H]−) (expected m/z, 1013.17. C60H62NO18P requires M, 1115.37).Dec-9-enyl 2,3,4-tri-O-benzoyl-α-D-fucopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-α-D-mannopyranosyl phosphate, triethylammonium salt 44
This compound was prepared by condensation of the H-phosphonate 43 (100 mg, 0.09 mmol) and dec-9-en-1-ol (0.035 cm3, 0.197 mmol) in pyridine (1 cm3) in the presence of trimethylacetyl chloride (0.03 cm3, 0.246 mmol) followed by oxidation with iodine (50 mg, 0.197 mmol) in pyridine–water (95∶5; 2 cm3) as described for the synthesis of the phosphodiester 15. FCC [CH2Cl2–MeOH–Et3N, (99∶0∶1) → (87∶12∶1)] gave the phosphodiester 44 (95 mg, 78%) as an amorphous solid, [α]26D +46 (c 0.99, CHCl3); δH (200 MHz) 1.04 (3 H, d, J5′,6′ 6.3, 6′-H3), 1.23 (10 H, m, 5 × CH2), 1.31 (9 H, t, 3 × MeCH2), 1.64 (2 H, tt, J 6.9, OCH2CH2), 1.99 (2 H, dt, J 6.9, CH2CH2CH), 3.10 (6 H, q, 3 × MeCH2), 4.00 (2 H, m, OCH2CH2), 4.44 (1 H, q, 5′-H), 4.57–4.70 (2 H, m, 5-H and 6-Ha), 4.79 (1 H, J3,4 = J4,5 = 9.8, 4-H), 4.89 (2 H, br d, 6-Hb and HCHCH), 4.96 (1 H, dd, 2JH,H 1.9, 3JH,H-E 17.0, HCHCH), 5.65–5.83 (8 H, m, 1-, 1′-, 2-, 2′-, 3-, 3′-, 4′-H and CH2CHCH2) and 7.00–8.25 (30 H, m, 6 × Ph); δP
−2.60; ESMS(−): m/z 1166.9 (100%, [M − Et3N − H]−) (expected m/z, 1167.31. C70H80NO19P requires M, 1269.51).
Dec-9-enyl α-D-fucopyranosyl-(1→4)-α-D-mannopyranosyl phosphate, triethylammonium salt 10
De-O-benzoylation of compound 44 (95 mg) with 0.05 mol dm−3 NaOMe in methanol (16 h at rt) followed by work-up, as described in the preparation of the phosphodiester 7, gave the phosphodiester 10 (48 mg, 100%) as an amorphous solid, [α]28D +69.8 (c 0.98, MeOH); δH (200 MHz; D2O) (inter alia) 1.23 (3 H, d, J5′,6′ 6.6, 6′-H3), 1.26-1.40 (19 H, m, 3 × MeCH2 and 5 × CH2), 1.61 (2 H, tt, J 6.5, OCH2CH2CH2), 2.04 (2 H, dt, J 7.0, CH2CH2CH), 3.20 (6 H, q, 3 × MeCH2), 5.20 (1 H, br s, 1′-H), 5.42 (1 H, br d, J1,P 7.6, 1-H) and 5.83 (1 H, m, CH2CHCH2); δC, δP and ESMS(−) data: see Table 1.
Acknowledgements
This work and I. A. I. were supported by a Wellcome Trust International Grant. The research of A. V. N. was supported by an International Research Scholar’s award from the Howard Hughes Medical Institute. One of us (A. J. R.) thanks the BBSRC for the award of a studentship.References
- Part 10 A. Crossman, Jr., J. S. Brimacombe, M. A. J. Ferguson and T. K. Smith, Carbohydr. Res., 1999, 321, 42 CrossRef CAS.
- M. J. McConville and M. A. J. Ferguson, Biochem. J., 1993, 294, 305 CAS.
- A. V. Nikolaev, T. J. Rutherford, M. A. J. Ferguson and J. S. Brimacombe, J. Chem. Soc., Perkin Trans. 1, 1995, 1977 RSC.
- A. V. Nikolaev, T. J. Rutherford, M. A. J. Ferguson and J. S. Brimacombe, J. Chem. Soc., Perkin Trans. 1, 1996, 1559 RSC.
- A. P. Higson, Y. E. Tsvetkov, M. A. J. Ferguson and A. V. Nikolaev, J. Chem. Soc., Perkin Trans. 1, 1998, 2587 RSC.
- A. P. Higson, Y. E. Tsvetkov, M. A. J. Ferguson and A. V. Nikolaev, Tetrahedron Lett., 1999, 40, 9281 CrossRef CAS.
- G. M. Brown, A. R. Millar, C. Masterson, J. S. Brimacombe, A. V. Nikolaev and M. A. J. Ferguson, Eur. J. Biochem., 1996, 242, 410 CrossRef CAS.
- I. A. Ivanova, A. J. Ross, M. A. J. Ferguson and A. V. Nikolaev, J. Chem. Soc., Perkin Trans. 1, 1997, 969 RSC.
- A. V. Nikolaev, I. A. Ivanova, V. N. Shibaev and N. K. Kochetkov, Carbohydr. Res., 1990, 204, 65 CrossRef CAS.
- N. S. Utkina, A. V. Nikolaev and V. N. Shibaev, Sov. J. Bioorg. Chem. (Engl. Transl.), 1991, 17, 303 Search PubMed.
- C. Copeland and R. V. Stick, Aust. J. Chem., 1978, 31, 1371 CAS.
- S. A. Abbas and A. H. Haines, Carbohydr. Res., 1975, 39, 358 CrossRef CAS.
- T. Nishio, Y. Miyake, K. Kubota, M. Yamai, S. Miki, T. Ito and T. Oku, Carbohydr. Res., 1996, 280, 357 CrossRef CAS.
- G. M. Bebault and G. G. S. Dutton, Can. J. Chem., 1972, 50, 3373 CAS.
- A. Hasegawa, K. Adachi, M. Yoshida and M. Kiso, Carbohydr. Res., 1992, 230, 273 CrossRef CAS.
- J. A. L. M. van Dorst, C. J. van Heusden, J. M. Tikkanen, J. P. Kamerling and J. F. G. Vliegenthart, Carbohydr. Res., 1997, 297, 209 CrossRef.
- K. Bock and K. Pedersen, Adv. Carbohydr. Chem. Biochem., 1983, 41, 27 Search PubMed.
- J. V. O’Conner, H. A. Nunez and R. Barker, Biochemistry, 1979, 18, 500 CrossRef.
- F. H. Routier, A. P. Higson, I. A. Ivanova, A. J. Ross, Y. E. Tsvetkov, D. V. Yashunsky, P. A. Bates, A. V. Nikolaev and M. A. J. Ferguson, Biochemistry, 2000, 39, 8017 CrossRef CAS.
Footnotes |
| † The value of 1JC1,H1 ≈ 170 Hz is typical for α-D-derivatives. For the β-D-glycosyl residues the value is about 160 Hz: for β-D-Galp in compound 7, 1JC1′,H1′ = 162.5 Hz (Table 1) (see also refs. 3, 4, 9 and 16). |
| ‡ For methyl β-D-altropyranoside, δC-5 = 75.60.17 |
| § For methyl β-D-rhamnopyranoside, δC-5 = 73.60.17 |
| This journal is © The Royal Society of Chemistry 2001 |