α-Chymotrypsin-catalysed peptide synthesis via the kinetically controlled approach using activated esters as acyl donors in organic solvents with low water content: incorporation of non-protein amino acids into peptides
Received
(in Cambridge, UK)
25th May 2000
, Accepted 30th October 2000
First published on 11th December 2000
Abstract
The coupling efficiency in the α-chymotrypsin-catalysed peptide synthesis via the kinetically controlled approach is greatly improved by the use of activated esters such as the 2,2,2-trifluoroethyl ester as acyl donors instead of the conventional methyl ester in organic solvents such as acetonitrile with low water content. This approach is useful for the incorporation of non-protein amino acids such as halogenophenylalanines into peptides.
Introduction
The use of proteases for peptide synthesis has attracted growing attention in recent years.1 Enzymic peptide synthesis has several advantages over conventional chemical methodologies: enzyme specificity suppresses side reactions and ensures the production of chemically and chirally pure peptides. Thus, peptide synthesis using enzymes has come to constitute an important complement to chemical methods and even to recombinant DNA techniques. However, most of the relevant works deal with peptides consisting of only proteinogenic amino acids which are universally distributed as protein constituents in all living organisms. Of hundreds of known amino acids, other naturally occurring ones are of non-protein origin,2 and there are also a number of purely synthetic (or unnatural) ones. Non-protein amino acids are important as building blocks for the synthesis of analogues of biologically active peptides necessary for structure–activity studies.3 The synthesis of peptides containing non-protein amino acids has usually been carried out by chemical methods, and moreover, recombinant DNA techniques are not easily applicable to the synthesis of peptides containing these non-coded amino acids. Therefore, the enzymic incorporation of non-protein amino acids into peptides remains a challenge.
We have recently investigated the optical resolution of non-protein amino acids![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 4via their ester hydrolysis using lipases5 and proteases6 from various sources, and consequently we have been interested in incorporating these amino acids into peptides using easily available proteases via the kinetically controlled approach.1a,b,e During the course of our investigation, we have found that the coupling efficiency can greatly be improved by the use of activated esters such as the 2,2,2-trifluoroethyl ester as an acyl donor instead of the conventional methyl ester.7 In this paper, we describe the details of our relevant works.
4via their ester hydrolysis using lipases5 and proteases6 from various sources, and consequently we have been interested in incorporating these amino acids into peptides using easily available proteases via the kinetically controlled approach.1a,b,e During the course of our investigation, we have found that the coupling efficiency can greatly be improved by the use of activated esters such as the 2,2,2-trifluoroethyl ester as an acyl donor instead of the conventional methyl ester.7 In this paper, we describe the details of our relevant works.
Results and discussion
Initially, we intended to use papain (EC 3.4.22.2), a sulfydryl protease from papaya latex which is known to possess a wide substrate specificity,1d for the purpose. Our recent study of the papain-catalysed enantioselective hydrolysis of non-protein amino acid esters also illustrated a rather broad substrate tolerance of this enzyme.6b We adopted the kinetically controlled approach where carboxy components were used in the form of esters at pH-values greater than 8, as introduced by Mitin et al.8 as opposed to the equilibrium-controlled approach. The methyl esters of N-Z derivatives of racemic non-protein amino acids were chosen as carboxy (C) components, because papain is known to exhibit a preference for bulky hydrophobic residues in the P2-position9 of the substrate, and they were allowed to react with L-Leu anilide used as an amino (N) component (Scheme 1). The use of racemic C-components would, if fortunate, allow peptide-bond formation and optical resolution simultaneously to yield homochiral peptides. The couplings were performed in Tris–maleate buffer (pH 8.0) at 35 °C with an N/C molar quotient of 1.5 which was determined through preliminary experiments. The progress of reaction was followed by reversed-phase HPLC analysis. In the couplings of amino acids carrying aliphatic side-chains as C-components, the targeted peptides were obtained generally in high yields, the coupling efficiency being reduced by branching in the side-chain and especially by a long side-chain of 7 or more carbon atoms.10 Fortunately, the diastereoselectivities toward the L-L peptides were sufficiently high. In sharp contrast, the yields and diastereoselectivities were low in the couplings of amino acids bearing aromatic side-chains as C-components (the first three entries in Table 1). Consequently, we turned our attention to the use of another protease, α-chymotrypsin (EC 3.4.21.1), which is known to hydrolyse preferentially a peptide bond adjacent to the carbonyl group of aromatic amino acid residues.1d Adopting the kinetically controlled approach in this case also, the methyl esters of N-Z-DL-halogenophenylalanines were treated with L-Leu anilide (N/C molar quotient 1.5) in carbonate–bicarbonate buffer (pH 10.0) containing 20% (v/v) DMF after some preliminary experiments searching for the optimum coupling conditions. As shown in Table 1, the yields of the targeted peptides were much higher than those obtained using papain as a catalyst. The replacement of a hydrogen by a halogen atom on the benzene ring caused a decrease in the coupling efficiency. This was especially serious with the coupling of the bromophenylalanine derivative. ortho-Substitution also caused a deterioration in the coupling yield. On the other hand, the diastereoselectivities toward the L-L peptides were high in these cases, with the exception of the p-fluorophenylalanine derivative.
Table 1 Protease-catalysed coupling of Z-DL-Phe(X)-OMea with L-Leu-NHPhb
| X |
Protease |
Yield (%)c |
% L-L |
|
Phe(X) stands for a Phe analogue bearing a substituent X on the benzene ring; for example, Phe(4F) represents 4-fluorophenylalanine.
Coupling conditions with α-chymotrypsin (α-CT): see the Experimental section. With papain: Z-DL-Phe(X)-OMe (0.5 mmol) and L-Leu-NHPh·HCl (182 mg, 0.75 mmol) were treated in the presence of papain (75 mg) and 2-mercaptoethanol (50 μl) in 7 ml of 0.05 M Tris–maleate buffer (pH 8.0) at 35 °C for 24 h.
Total yield of peptide diastereomers (L-L + D-L).
|
| H |
Papain |
8.1 |
57 |
| 4F |
Papain |
6.3 |
55 |
| 4Cl |
Papain |
5.9 |
54 |
| H |
α-CT |
39.2 |
93 |
| 4F |
α-CT |
29.1 |
86 |
| 2Cl |
α-CT |
25.8 |
99 |
| 4Cl |
α-CT |
35.0 |
97 |
| 4Br |
α-CT |
15.2 |
98 |
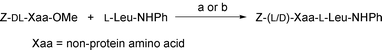 |
| | Scheme 1 Protease-catalysed couplings of non-protein DL-amino acids (DL-Xaa) as C-components. Enzyme and conditions: (a) papain, Tris–maleate buffer (pH 8.0), 35 °C; (b) α-chymotrypsin, carbonate–bicarbonate buffer (pH 10.0) containing 20% (v/v) DMF, 35 °C. | |
The protease-catalysed peptide-bond formation based on the kinetically controlled approach can be carried out in organic solvents with low water content as well as in aqueous media.11 It is expected that in the former the water activity is reduced, which favours the synthetic reaction and virtually eliminates the secondary hydrolysis of the peptide formed. In addition, our recent experiences with the successful use of 2-chloroethyl and 2,2,2-trifluoroethyl esters in the lipase-catalysed enantioselective hydrolysis5a,b or transesterification5c of amino ester substrates prompted us to try such activated esters as acyl donors instead of the conventional methyl ester.7 Along these lines, we chose the α-chymotrypsin-catalysed synthesis of Z-L-Phe-L-Leu-NH2 as a model system and several kinds of activated esters were evaluated as donor esters (Scheme 2). Hydrophilic organic solvents such as acetonitrile, propan-1-ol, 2-methylbutan-2-ol, acetone, 1,4-dioxane, THF, etc. containing a small amount of water were tested, and acetonitrile was found to be the best. Table 2 shows the results (after 1 h of incubation) of couplings conducted in acetonitrile containing different amounts [2–5% (v/v)] of Tris buffer (pH 7.8)12 in the presence of the enzyme adsorbed on Celite. In this case, not only the yield of the desired peptide but also that of the hydrolysis product of the donor ester was determined by HPLC analysis. Under the present reaction conditions, non-enzymic reactions (peptide-bond formation and hydrolysis of the donor ester) were not detected even when the activated esters were employed. The optimal water content was found to be 4%, and the solvent of this composition was used throughout further experiments. The coupling yield was almost doubled when the acyl donor was switched from the methyl ester to the 2-chloroethyl ester,13 and the use of the 2,2,2-trifluoroethyl ester14 was much more effective, forcing the reaction to completion, while the competing hydrolysis of the donor ester was little accelerated compared with the case of the methyl ester. Such a significant increase in the coupling efficiency on replacing the methyl ester with the trifluoroethyl ester was also observed in the couplings with several other amino acid amides as nucleophiles.7
| |
|
Yield (%)b |
| R |
Water content(%, v/v) |
Peptide |
Z-L-Phe-OH |
|
A mixture of Z-L-Phe-OR (0.05 mmol), L-Leu-NH2·HCl (0.2 mmol), TEA (0.2 mmol), and the immobilised enzyme (150 mg) was incubated with shaking in a solvent composed of 2 ml of acetonitrile and 41 μl (water content: 2%), 62 μl (3%), 83 μl (4%), or 105 μl (5%) of 0.05 M Tris buffer (pH 7.8) at 30 °C.
After 1 h of incubation.
|
| Me |
2 |
20.2 |
0.7 |
| |
3 |
26.9 |
1.0 |
| |
4 |
31.0 |
1.1 |
| |
5 |
31.3 |
1.3 |
| CH2CF3 |
2 |
87.1 |
1.8 |
| |
3 |
93.7 |
2.2 |
| |
4 |
98.2 |
2.3 |
| |
5 |
98.1 |
2.2 |
| CH2CH2Cl |
4 |
56.8 |
1.4 |
| CH2CCl3 |
4 |
96.3 |
2.5 |
| CH(CF3)2 |
4 |
70.2 |
1.9 |
 |
| | Scheme 2 | |
This promising method employing the trifluoroethyl ester was next applied to the couplings of non-protein amino acids as C-components in acetonitrile containing 4% Tris buffer (Scheme 3). Table 3 shows a comparison between the methyl and trifluoroethyl esters in the couplings of N-Z-DL-halogenophenylalanines with L-Leu-NH2. The peptide yields obtained using methyl esters were rather low; especially noteworthy was the deleterious effect of bulky substituents on the aromatic ring (o-Cl or o-Br). On the other hand, the yields were greatly improved by the use of trifluoroethyl esters without a large increase in the yields of hydrolysis products of the donor esters. In some cases, the reaction proceeded almost to completion. Moreover, it is worth noting that the reactions proceeded diastereospecifically to produce only the L-L peptides even when the trifluoroethyl esters were employed.
| |
|
Yield (%)b |
| X |
R |
Peptidec |
Z-Phe(X)-OH |
|
Coupling conditions as described in the Experimental section, using 0.1 mmol of Z-DL-Phe(X)-OR, 0.2 mmol of L-Leu-NH2·HCl, 0.2 mmol of TEA, and 150 mg of the immobilised enzyme in a solvent composed of 2 ml of acetonitrile and 83 μl of 0.05 M Tris buffer (pH 7.8) at 30 °C.
After 1 h of incubation.
Only the L-L peptide was detected in each case.
|
| 2F |
Me |
15.9 |
3.9 |
| |
CH2CF3 |
45.6 |
2.2 |
| 3F |
Me |
15.3 |
1.6 |
| |
CH2CF3 |
47.5 |
2.3 |
| 4F |
Me |
19.2 |
1.5 |
| |
CH2CF3 |
48.7 |
1.2 |
| 2Cl |
Me |
3.2 |
1.0 |
| |
CH2CF3 |
19.2 |
3.2 |
| 3Cl |
Me |
21.3 |
2.8 |
| |
CH2CF3 |
45.7 |
3.2 |
| 4Cl |
Me |
3.9 |
1.2 |
| |
CH2CF3 |
48.3 |
1.6 |
| 2Br |
Me |
0.8 |
0.5 |
| |
CH2CF3 |
13.9 |
3.4 |
| 3Br |
Me |
18.8 |
3.0 |
| |
CH2CF3 |
42.4 |
3.6 |
| 4Br |
Me |
1.4 |
0.2 |
| |
CH2CF3 |
40.4 |
2.2 |
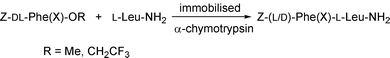 |
| | Scheme 3 Immobilised α-chymotrypsin-catalysed couplings of DL-halogenophenylalanines as C-components. Conditions: acetonitrile containing 4% (v/v) Tris buffer (pH 7.8), 30 °C. | |
The reaction mechanism of the α-chymotrypsin-catalysed peptide-bond formation via the kinetically controlled approach studied here is thought to be the one described for the synthesis of Z-L-Phe-L-Leu-NH2 as an example in Scheme 4.1b,e The ester substrate acting as an acyl donor first forms the enzyme–substrate (ES) complex with the enzyme, and then it affords the acyl-enzyme intermediate with the liberation of the leaving group (OR). This intermediate is then deacylated either by a nucleophilic amino component to yield the peptide product or by water to form the hydrolysis product of the donor ester. The results obtained in this study indicate the usefulness of the activated esters such as the 2,2,2-trifluoroethyl ester as acyl donors. Since there seems to be no significant difference in the binding of the donor ester onto the enzyme between the methyl and trifluoroethyl esters, for example, the acylation rates of the enzyme should be very different between them: the activated ester must accelerate the acylation step compared with the conventional methyl ester. The successful incorporation of non-protein amino acids such as halogenophenylalanines into peptides illustrated in this paper suggests that the present approach will be promising for broadening the narrow substrate tolerance of enzymes, which will be the subject of the following paper.
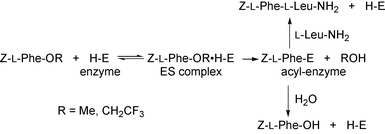 |
| | Scheme 4 | |
Experimental
All solvents were distilled and dried over molecular sieves prior to use. Amino acid amide hydrochlorides were purchased from Kokusan Chemical Works (Japan). Papain (twice crystallised, lyophilised powder, ex papaya latex) was purchased from Sigma and had a specific activity of 2.8 units per mg solid with Bz-L-Arg-OEt. α-Chymotrypsin (type II, ex bovine pancreas) was also from Sigma and had a specific activity of 48 units per mg solid with Bz-L-Tyr-OEt. IR spectra were recorded on a Nicolet N-750B FT-IR spectrometer using attenuated total reflectance (ATR). Wavenumbers of only the peaks ascribed to the N–H and C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O stretching vibrations are quoted. 1H NMR spectra (300 MHz) were recorded with a Varian Unity 300 spectrometer using CDCl3 as solvent with TMS as internal standard unless otherwise stated. J-Values are given in Hz. Mps were determined on a Yamato (Japan) MP-21 apparatus and are uncorrected. Optical rotations were measured with a JASCO (Japan) DIP-4 digital polarimeter, with [α]D-values given in units of 10−1 deg cm2 g−1.
O stretching vibrations are quoted. 1H NMR spectra (300 MHz) were recorded with a Varian Unity 300 spectrometer using CDCl3 as solvent with TMS as internal standard unless otherwise stated. J-Values are given in Hz. Mps were determined on a Yamato (Japan) MP-21 apparatus and are uncorrected. Optical rotations were measured with a JASCO (Japan) DIP-4 digital polarimeter, with [α]D-values given in units of 10−1 deg cm2 g−1.
Preparation of Z-L- or Z-DL-Phe(X)
L- or DL-Phe(X) (X = H, 2F, 3F, 4F, 2Cl, 3Cl, 4Cl, 2Br, 3Br or 4Br) was benzyloxycarbonylated using Z-Cl under the usual Schotten–Baumann conditions15 to give Z-L- or Z-DL-Phe(X), which was purified by recrystallisation from an appropriate solvent [e.g., EtOAc–petroleum spirit (bp 30–70 °C)]. The physical properties of Z-DL-Phe(X) bearing X = 2F, 3F, 4F and 4Cl were reported elsewhere.5d The mps (°C) of the other Z-DL-Phe(X) employed in this study are as follows: X = 2Cl, 124–125; X = 3Cl, 92.5–93; X = 2Br, 126–127; X = 3Br, 99–99.5; X = 4Br, 166–168.
Preparation of Z-L- or Z-DL-Phe(X)-OMe
Treatment of Z-L- or Z-DL-Phe(X) with an ethereal solution of diazomethane afforded their methyl esters in a nearly quantitative yield. Z-L-Phe-OMe, oil; [α]25D
−14.4 (c 1.3, MeOH) {lit.,16 [α]19D
−15.6 (c 1.02, MeOH)}. The mps (°C) of Z-DL-Phe(X)-OMe obtained as crystals are as follows: X = 3F, 56–58; X = 2Cl, 66.5–68; X = 4Cl, 68–70; X = 4Br, 86–88. The methyl esters bearing X = 2F, 4F and 2Br were obtained as oils.
Preparation of the halogenated alkyl esters of Z-L- or Z-DL-Phe(X)
Z-L- or Z-DL-Phe(X) was converted to its halogenated alkyl esters by treatment with the corresponding halogenated alcohol in the presence of DMAP and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) in DCM according to the procedure of Dhaon et al.17 The preparation of Z-L-Phe-OCH2CF3 is described as a typical example. To a stirred solution of Z-L-Phe (660 mg, 2.2 mmol), DMAP (135 mg, 1.1 mmol) and 2,2,2-trifluoroethanol (250 mg, 2.5 mmol) in dry DCM (8 ml) was added EDC·HCl (460 mg, 2.4 mmol) under ice-cooling, and the mixture was stirred at this temperature for 2 h and then at ambient temperature overnight. The reaction mixture was partitioned between EtOAc and water, and the organic layer was washed successively with 1 M HCl, water, 1 M aq. NaHCO3 and brine, and dried over Na2SO4. Evaporation of the solvent in vacuo gave crystals in 96% yield; mp 80–80.5 °C; [α]25D
−20.8 (c 1.0, MeOH) (Found: C, 59.90; H, 4.73; N, 3.79. C19H18F3NO4 requires C, 59.84; H, 4.76; N, 3.67%); νmax/cm−1 3324, 1764, 1686; δH 3.06–3.22 (2H, d of AB q, J 15.3 and 6.6), 4.37–4.61 (2H, m), 4.76 (1H, m), 5.10 (2H, apparent s), 5.16 (1H, br d, J 7), 7.10–7.36 (5H, m), 7.34 (5H, apparent s).
Z-L-Phe-OCH2CH2Cl..
83% yield; mp 43–44 °C; [α]25D
−20.3 (c 1.0, MeOH) (Found: C, 63.17; H, 5.52; N, 3.98. C19H20ClNO4 requires C, 63.07; H, 5.57; N, 3.87%); νmax/cm−1 3346, 1752, 1683; δH 3.13 (2H, m), 3.61 (2H, t, J 5.7), 4.34 (2H, m), 4.69 (1H, m), 5.00 (1H, apparent s), 5.23 (1H, br d, J 8), 7.11–7.38 (5H, m), 7.33 (5H, apparent s).
Z-L-Phe-OCH2CCl3..
92% yield; oil; [α]25D +6.9 (c 1.3, CHCl3) (Found: C, 53.11; H, 4.20; N, 3.24. C19H18Cl3NO4 requires C, 52.98; H, 4.21; N, 3.25%); νmax/cm−1 3333, 1704; δH 3.09–3.28 (2H, d of AB q, J 14.1 and 6.9), 4.75 (2H, AB q, J 12.0), 4.81 (1H, m), 5.00 (1H, apparent s), 5.18 (1H, br d, J 8), 7.14–7.38 (5H, m), 7.33 (5H, apparent s).
Z-L-Phe-OCH(CF3)2..
95% yield; mp 73.5–75 °C; [α]25D +3.2 (c 1.1, CHCl3) (Found: C, 53.78; H, 3.78; N, 3.26. C20H17F6NO4 requires C, 53.46; H, 3.81; N, 3.12%); νmax/cm−1 3327, 1782, 1686; δH 3.05–3.26 (2H, d of AB q, J 14.1 and 6.5), 4.83 (1H, m), 5.09 (2H, apparent s), 5.12 (1H, br d, J 7), 5.77 (1H, septet, J 6.0), 7.11–7.39 (10H, m).
Z-DL-Phe(2F![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) )-OCH2CF3..
84% yield; mp 39–40 °C (Found: C, 56.97; H, 4.21; N, 3.27. C19H17F4NO4 requires C, 57.15; H, 4.29; N, 3.51%); νmax/cm−1 3331, 1772, 1696; δH 3.09–3.31 (2H, m), 4.49 (2H, q, J 8.4), 4.75 (1H, m), 5.09 (2H, AB q, J 12.3), 5.25 (1H, br d, J 9), 7.00–7.39 (9H, m).
)-OCH2CF3..
84% yield; mp 39–40 °C (Found: C, 56.97; H, 4.21; N, 3.27. C19H17F4NO4 requires C, 57.15; H, 4.29; N, 3.51%); νmax/cm−1 3331, 1772, 1696; δH 3.09–3.31 (2H, m), 4.49 (2H, q, J 8.4), 4.75 (1H, m), 5.09 (2H, AB q, J 12.3), 5.25 (1H, br d, J 9), 7.00–7.39 (9H, m).
Z-DL-Phe(3F)-OCH2CF3..
94% yield; mp 68–69 °C (Found: C, 57.31; H, 4.02; N, 3.56%); νmax/cm−1 3318, 1762, 1685; δH 3.05–3.22 (2H, d of AB q, J 14.1 and 5.4), 4.40–4.60 (2H, m), 4.73 (1H, m), 5.10 (2H, AB q, J 6.2), 5.21 (1H, br d, J 8), 6.80–7.39 (4H, m), 7.35 (5H, apparent s).
Z-DL-Phe(4F)-OCH2CF3..
91% yield; mp 79–81 °C (Found: C, 57.32; H, 4.37; N, 3.30%); νmax/cm−1 3328, 1761, 1687; δH 3.03–3.20 (2H, d of AB q, J 14.1 and 6.6), 4.37–4.62 (2H, m), 4.75 (1H, m), 5.13 (2H, AB q, J 14.1), 5.19 (1H, br d, J 8), 6.97–7.40 (9H, m).
Z-DL-Phe(2Cl)-OCH2CF3..
91% yield; mp 79–80 °C (Found: C, 55.03; H, 4.09; N, 3.62. C19H17ClF3NO4 requires C, 54.88; H, 4.12; N, 3.37%); νmax/cm−1 3352, 1766, 1696; δH 3.15–3.38 (2H, d of AB q, J 14.1 and 6.3), 4.49 (2H, q, J 8.4), 4.79 (1H, m), 5.07 (2H, AB q, J 12.9), 5.26 (1H, br d, J 8), 7.16–7.39 (9H, m).
Z-DL-Phe(3Cl)-OCH2CF3..
86% yield; mp 66–67 °C (Found: C, 54.69; H, 4.36; N, 3.09%); νmax/cm−1 3321, 1766, 1689; δH (DMSO-d6) 2.92 (1H, dd-like, J 13.8 and 5.4), 3.07 (1H, dd-like, J 13.8 and 5.1), 4.41 (1H, m), 4.78 (2H, q, J 9.0), 4.99 (2H, AB q, J 12.6), 7.21–7.37 (9H, m), 7.99 (1H, d, J 7.8).
Z-DL-Phe(4Cl)-OCH2CF3..
85% yield; mp 95–101 °C (Found: C, 55.16; H, 4.27; N, 3.39%); νmax/cm−1 3343, 1766, 1697; δH 3.02–3.20 (2H, d of AB q, J 14.1 and 6.0), 4.37–4.63 (2H, m), 4.74 (1H, m), 5.09 (2H, AB q, J 12.3), 5.16 (1H, br d, J 8), 7.02–7.40 (9H, m).
Z-DL-Phe(2Br)-OCH2CF3..
95% yield; mp 83.5–84 °C (Found: C, 49.50; H, 4.01; N, 3.27. C19H17BrF3NO4 requires C, 49.58; H, 3.72; N, 3.04%); νmax/cm−1 3320, 1770, 1697; δH 3.14–3.41 (2H, m), 4.45–4.55 (2H, m), 4.80 (1H, m), 5.06 (2H, s), 5.27 (1H, br d, J 9), 7.09–7.57 (9H, m).
Z-DL-Phe(3Br)-OCH2CF3..
88% yield; mp 76–77 °C (Found: C, 49.81; H, 3.67; N, 3.24%); νmax/cm−1 3319, 1766, 1689; δH (DMSO-d6) 2.90 (1H, dd-like, J 12.0 and 10.5), 3.07 (1H, dd-like, J 12.0 and 4.8), 4.41 (1H, m), 4.77 (2H, q, J 9.0), 4.98 (2H, AB q, J 12.3), 7.20–7.50 (9H, m), 7.98 (1H, d, J 7.8).
Z-DL-Phe(4Br)-OCH2CF3..
85% yield; mp 111–113 °C (Found: C, 49.44; H, 3.59; N, 3.25%); νmax/cm−1 3322, 1761, 1687; δH 3.00–3.19 (2H, d of AB q, J 14.1 and 6.3), 4.37–4.63 (2H, m), 4.74 (1H, m), 5.09 (2H, AB q, J 12.3), 5.16 (1H, br d, J 8), 6.97–7.43 (9H, m).
Preparation of L-Leu-NHPh·HCl
Boc-L-Leu·1/2H2O (2.4 g, 10 mmol), aniline (1.4 g, 15 mmol), EDTA (60 mg) and L-Cys (200 mg) were dissolved in a mixture of pH 4.8 acetate buffer (140 ml) and DMF (60 ml). After the pH had been adjusted to 4.8, papain (400 mg) was added and the mixture was stirred at 38 °C for 4 days. The precipitate was filtered off, washed with the buffer, and dried in a vacuum desiccator over P4O10. Materials insoluble in EtOAc were filtered off and the filtrate was concentrated to give white crystals (2.6 g; mp 136–143 °C), which were recrystallised from EtOAc–petroleum spirit to give Boc-L-Leu-NHPh (1.7 g, 56%); mp 148–150 °C; [α]25D
−34.8 (c 1.0, MeOH) (Found: C, 70.12; H, 9.28; N, 9.63. C17H26N2O2 requires C, 70.31; H, 9.02; N, 9.65%). Boc-L-Leu-NHPh (1.5 g, 5 mmol) thus obtained was stirred in 3.7 M HCl in 1,4-dioxane (15 ml) for 1.5 h. Evaporation of the solvent in vacuo afforded white crystals, which were recrystallised from propan-2-ol–diisopropyl ether to give L-Leu-NHPh·HCl (0.9 g, 74%); mp 218–222 °C.
Preparation of authentic dipeptides
An authentic sample of Z-L-Phe-L-Leu-NH2 was prepared as follows. To a stirred solution of Z-L-Phe (300 mg, 1.0 mmol), L-Leu-NH2·HCl (130 mg, 1.0 mmol), triethylamine (TEA) (100 mg, 1.0 mmol) and HOBT (135 mg, 1.0 mmol) in DMF (10 ml) was added EDC·HCl (190 mg, 1.0 mmol) under ice-cooling. After stirring at this temperature for 2 h and then at ambient temperature overnight, the reaction mixture was diluted with EtOAc, washed successively with 1 M HCl, water, 1 M aq. NaHCO3 and brine, and dried over Na2SO4. Evaporation of the solvent in vacuo afforded crystals, which were recrystallised from aq. MeOH (370 mg, 90%); mp 192.5–193 °C; [α]25D
−18.3 (c 1.0, DMF), −21.7 (c 1.0, MeOH) {lit.,18 mp 195–196 °C; [α]25D
−19.7 (c 1, DMF)}; δH (DMSO-d6) 0.83 (3H, d, J 6.3), 0.87 (3H, d, J 6.6), 1.45 (2H, apparent t, J 7), 1.48–1.66 (1H, m), 2.72 (1H, dd, J 13.8 and 11.1), 3.00 (1H, dd, J 13.8 and 3.9), 4.16–4.33 (2H, m), 4.93 (2H, AB q, J 13.2), 6.99 (1H, s), 7.11–7.35 (11H, m), 7.51 (1H, d, J 8.4), 7.97 (1H, d, J 8.4).
The mixtures of diastereomers (L-L and D-L) of Z-Phe(X)-Leu-NHPh or Z-Phe(X)-Leu-NH2 were prepared through the coupling of Z-DL-Phe(X) with L-Leu-NHPh or L-Leu-NH2 (in the form of the hydrochloride with an equimolar amount of TEA) by the EDC–HOBT method in DMF in the same manner as above and purified by recrystallisation from aq. MeOH. Each sample thus prepared showed only two peaks, corresponding to both the diastereomers on reversed-phase HPLC. The faster eluting diastereomer proved to be L-L by comparison with authentic samples prepared through the coupling of a very small amount of Z-L-Phe(X) (X = 2F, 3F, 4F, 4Cl and 4Br), which was obtained by enzymic resolution,5,6 with L-Leu-NHPh or L-Leu-NH2 salt by the EDC–HOBT method.
Preparation of immobilised α-chymotrypsin
α-Chymotrypsin (32 mg) was dissolved in 4 ml of 0.05 M Tris buffer (pH 7.8) and mixed with Celite No. 535 (Johns-Mansville Co., Denver, CO; 1 g), and the mixture was lyophilised using an Eyela (Japan) freeze-dryer FDU-830 for 20 h.
HPLC analyses
The liquid chromatograph employed was a GL Sciences (Japan) PU-610 instrument, equipped with a Rheodyne 8125 sample injector and a GL Sciences UV-620 variable-wavelength UV monitor. A Shimadzu (Japan) C-R6A data processor was used for data acquisition and processing. The amounts of the donor ester, peptide and hydrolysis product of the donor ester were determined by HPLC analysis on an ODS column under the following conditions: column, Inertsil ODS 3 (5 μm; 1.5 mm id × 150 mm, GL Sciences); mobile phase, 55–60% aq. MeOH containing H3PO4 (0.01 M); flow rate, 0.1 ml min−1; column temperature, 40 °C; detection, UV at 254 nm. The diastereomers (L-L and D-L) of the resulting peptides were also separated well on the same column by decreasing the amount of MeOH in the mobile phase. The separation of the diastereomers (L-L and D-L) of Z-Phe(X)-Leu-NH2 is compiled in Table 4.
Table 4 HPLC separation of the diastereomers (L-L and D-L) of Z-Phe(X)-Leu-NH2a
| X |
Mobile phaseb |
k′L-Lc |
k′D-Lc |
αd |
|
HPLC conditions: see the Experimental section.
A, 55% aq. MeOH containing H3PO4 (0.01 M); B, 60% aq. MeOH containing H3PO4 (0.01 M).
Capacity factor: k′ = (tR − t0)/t0, where t0 = 1.50 min.
Separation factor: α = k′D-L/k′L-Lp.
|
| 2F |
A |
16.5 |
20.1 |
1.22 |
| 3F |
A |
18.8 |
22.4 |
1.19 |
| 4F |
A |
18.8 |
21.5 |
1.14 |
| 2Cl |
B |
12.9 |
16.2 |
1.26 |
| 3Cl |
B |
15.0 |
18.1 |
1.21 |
| 4Cl |
B |
16.3 |
18.3 |
1.12 |
| 2Br |
B |
14.1 |
17.9 |
1.27 |
| 3Br |
B |
17.9 |
21.7 |
1.21 |
| 4Br |
B |
19.5 |
21.5 |
1.10 |
α-Chymotrypsin-catalysed coupling of Z-DL-Phe(X)-OMe with L-Leu-NHPh in aqueous media
Z-DL-Phe(X)-OMe (0.25 mmol) and L-Leu-NHPh·HCl (92 mg, 0.38 mmol) were added to a mixture of 5 ml of carbonate–bicarbonate buffer (pH 10.0) and 1.2 ml of DMF, followed by α-chymotrypsin (32 mg). The reaction mixture was stirred at 35 °C for 24 h. After addition of 10% (w/v) aq. citric acid (10 ml), the mixture was extracted with EtOAc. After addition of p-nitrotoluene (13.7 mg, 0.1 mmol) as internal standard, the combined EtOAc extracts were washed successively with 1 M HCl, 5% aq. NaHCO3, and brine, and dried over Na2SO4. After evaporation of the solvent, a part of the residue was subjected to reversed-phase HPLC analysis using aq. MeOH as the mobile phase to obtain the coupling yield. Calibration plots of the ratio of peak areas on reversed-phase HPLC between each targeted peptide (synthesised chemically as a mixture of diastereomers) and p-nitrotoluene against their molar ratio gave straight lines going through the origin, which were used for estimating the coupling yield.
Peptide synthesis mediated by immobilised α-chymotrypsin in acetonitrile with low water content
The coupling of Z-DL-Phe(2F)-OCH2CF3 with L-Leu-NH2 is described as a typical example. A mixture of Z-DL-Phe(2F)-OCH2CF3 (40 mg, 0.1 mmol), L-Leu-NH2·HCl (33 mg, 0.2 mmol), TEA (28 μl, 0.2 mmol) and the immobilised enzyme (150 mg, corresponding to 4.7 mg of α-chymotrypsin) was incubated with shaking (180 strokes min−1) in a solvent composed of 2 ml of acetonitrile and 83 μl of 0.05 M Tris buffer (pH 7.8) at 30 °C. The amounts of the donor ester, peptide and hydrolysis product were determined as described in ‘HPLC analyses,’ above.
Acknowledgements
This work was supported in part by grants from the Science Research Promotion Fund of the Japan Private School Promotion Foundation and from the Ministry of Education, Science and Culture, Japan (to T. M.).
References
- For reviews, see:
(a)
K. Faber, Biotransformations in Organic Chemistry, Springer-Verlag, Berlin, 3rd edn., 1997, p. 331;
Search PubMed;
(b)
H.-D. Jakubke, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz and H. Waldmann, VCH, Weinheim, 1995, B.2.5, p. 431;
Search PubMed;
(c)
C.-H. Wong and G. M. Whitesides, Enzymes in Synthetic Organic Chemistry, Pergamon Press, Oxford, 1994, p. 46;
Search PubMed;
(d)
W. Kullmann, Enzymatic Peptide Synthesis, CRC Press, Boca Raton, FL, 1987;
Search PubMed;
(e)
H.-D. Jakubke, in The Peptides, ed. S. Udenfriend and J. Meienhofer, Academic Press, San Diego, 1987, vol. 9, ch. 3.
Search PubMed.
-
S. Hunt, in Chemistry and Biochemistry of the Amino Acids, ed. G. C. Barrett, Chapman and Hall, London, 1985, p. 55.
Search PubMed.
-
D. C. Roberts and F. Vellaccio, in The Peptides, ed. E. Gross and J. Meienhofer, Academic Press, New York, 1983, vol. 5, ch. 6.
Search PubMed.
- For reviews, see:
(a) T. Miyazawa, Amino Acids, 1999, 16, 191 CAS;
(b)
T. Miyazawa, in Fluorine-containing Amino Acids: Synthesis and Properties, ed. V. P. Kukhar’ and V. A. Soloshonok, Wiley, Chichester, 1995, p. 267.
Search PubMed.
-
(a) T. Miyazawa, T. Takitani, S. Ueji, T. Yamada and S. Kuwata, J. Chem. Soc., Chem. Commun., 1988, 1214 RSC;
(b) T. Miyazawa, H. Iwanaga, S. Ueji, T. Yamada and S. Kuwata, Chem. Lett., 1989, 2219 CAS;
(c) T. Miyazawa, M. Mio, Y. Watanabe, T. Yamada and S. Kuwata, Biotechnol. Lett., 1992, 14, 789 CrossRef CAS;
(d) T. Miyazawa, H. Iwanaga, S. Ueji and T. Yamada, Biocatal. Biotransform., 2000, 17, 445 Search PubMed.
-
(a) T. Miyazawa, H. Iwanaga, T. Yamada and S. Kuwata, Chirality, 1992, 4, 427 CrossRef CAS;
(b) T. Miyazawa, H. Iwanaga, T. Yamada and S. Kuwata, Biotechnol. Lett., 1994, 16, 373 CrossRef CAS;
(c) T. Miyazawa, H. Minowa, T. Miyamoto, K. Imagawa, R. Yanagihara and T. Yamada, Tetrahedron: Asymmetry, 1997, 8, 367 CrossRef CAS;
(d) T. Miyazawa, K. Imagawa, R. Yanagihara and T. Yamada, Biotechnol. Tech., 1997, 11, 931 CrossRef CAS.
- Preliminary communication T. Miyazawa, S. Nakajo, M. Nishikawa, K. Imagawa, R. Yanagihara and T. Yamada, J. Chem. Soc., Perkin Trans. 1, 1996, 2867 RSC.
- Yu. V. Mitin, N. P. Zapevalova and E. Yu. Gorbunova, Int. J. Pept. Protein Res., 1984, 23, 528 Search PubMed.
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157 CAS.
- Preliminary communication T. Miyazawa, K. Hamahara, M. Matsuoka, Y. Shindo and T. Yamada, Biotechnol. Lett., 1998, 20, 389 CrossRef CAS.
-
cf.
P. Clapés, P. Adlercreutz and B. Mattiasson, Biotechnol. Appl. Biochem., 1990, 12, 376 Search PubMed.
- The optimal pH for amide-bond hydrolysis mediated by α-chymotrypsin: see ref. 1(d)..
- This ester was used as an acyl donor in the incorporation of D-amino acids into peptides mediated by subtilisin in 2-methylbutan-2-ol: A. L. Margolin, D.-F. Tai and A. M. Klibanov, J. Am. Chem. Soc., 1987, 109, 7885 CrossRef CAS.
- 2,2,2-Trifluoroethyl esters were employed in the protease-catalysed transesterifications, see: B. Danieli, P. Bellis, G. Carrea and S. Riva, Gazz. Chim. Ital., 1991, 121, 123 Search PubMed; J. Broos, J. F. J. Engbersen, I. K. Sakodinskaya, W. Verboom and D. N. Reinhoudt, J. Chem. Soc., Perkin Trans. 1, 1995, 2899 RSC; H. G. Park, H. N. Chang and J. S. Dordick, Biotechnol. Lett., 1995, 17, 1085 CrossRef CAS; K. Kawashiro, H. Sugahara, S. Sugiyama and H. Hayashi, Biotechnol. Bioeng., 1997, 53, 26 CrossRef CAS.
-
M. Bodanszky and A. Bodanszky, The Practice of Peptide Synthesis, Springer-Verlag, Berlin, 2nd edn., 1994, p. 11.
Search PubMed.
- A. Ito, R. Takahashi, C. Miura and Y. Baba, Chem. Pharm. Bull., 1975, 23, 3106 Search PubMed.
- M. K. Dhaon, R. K. Olsen and K. Ramasamy, J. Org. Chem., 1982, 47, 1962 CrossRef CAS.
- P. Kuhl, U. Zacharias, H. Burckhardt and H.-D. Jakubke, Monatsh. Chem., 1986, 117, 1195 CAS.
|
| This journal is © The Royal Society of Chemistry 2001 |
Click here to see how this site uses Cookies. View our privacy policy here.