
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting oxime–nitrone tautomerism. Evidence of nitrone tautomer participation in oxime nucleophilic addition reactions†
David
Roca-López
,
Andrea
Darù
,
Tomás
Tejero
and
Pedro
Merino
*
Laboratorio de Síntesis Asimétrica, Departamento de Síntesis y Estructura de Biomoléculas, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), Universidad de Zaragoza, CSIC, Campus San Francisco, E-50009 Zaragoza, Aragón, Spain. E-mail: pmerino@unizar.es
First published on 19th February 2016
Abstract
The oxime–nitrone tautomerism has been revisited using high-level DFT calculations. The isomerization has been found to be more favorable through a bimolecular process involving two molecules of oxime, a finding that argues against the commonly accepted thermal 1,2-H-shift mechanism. The reaction of arylamidoximes with 1,2-diaza-1,3-dienes to yield the corresponding O-substituted oximes (stable intermediates for the synthesis of 1,2,4-oxadiazine derivatives) was also investigated as a rare case in which O-alkylation is observed in the reaction between oximes and electron-poor alkenes in the absence of a base. Under such conditions the reaction usually proceeds through the nucleophilic attack of the oxime nitrogen to yield the corresponding nitrone. The computational investigation revealed that in the case of arylamidoximes, the pathway involving the less stable but more reactive nitrone tautomer is the predominant mechanism, evidencing for the first time the involvement of a nitrone tautomer in nucleophilic additions of oximes. Validation of the model has been carried out by studying alternative ene-like processes; the dramatically different reactivity predicted for arylamidoximes and unsubstituted oxime are rationalized in terms of steric hindrance.
Introduction
About 40 years ago, Vijfhuizen and Terlouw reported the first evidence, observed by mass spectrometry, of the oxime–nitrone isomerization (Scheme 1).1 Two years later, Dignam and Hegarty studied isomerization of oximes in aqueous solution as a function of the pH and concluded that at high pH values the reactive species is the neutral oxime rather than the nitrone tautomer.2 In 1984, Grigg and co-workers proposed that isomerization of oxime to nitrone consisted of a thermal 1,2-hydrogen shift.3 | ||
| Scheme 1 Oxime–nitrone tautomerism. | ||
Since these early reports, cycloaddition reactions of oximes towards isoxazolidines have been extensively studied.4 In particular, the intramolecular version of the reaction (the so-called “intramolecular oxime olefin cycloaddition”, IOOC) has received considerable attention5 in contrast to intermolecular reactions which have been less studied.6 In this context, Tamura and co-workers developed intra-7 and intermolecular8 cycloadditions of O-silylated oximes that required milder conditions by using boron trifluoride etherate as a promoter of the reaction, leading to N-boranonitrones. Frank and co-workers reported a combined experimental and computational study demonstrating that even though the nitrone tautomer is less stable than the oxime one, the former is more reactive, leading to more stable transition structures.9 All these studies concern cycloaddition reactions of the nitrone tautomer. However, there are no reports illustrating the participation of such a tautomer in nucleophilic additions in spite of the fact that nitrones can act as excellent electrophiles.10
The mechanism of the isomerization has been discussed in various studies,11 all of them claiming for the previously proposed 1,2-hydrogen shift. Noguchi and co-workers12 found a dependence on the type of solvent but no evidences on the effect of alcoholic solvents. The authors suggested a solvation effect favoring the isomerization towards nitrone. The influence of substituents in the tautomerism has also been considered13 and in some particular case, a facile isomerization has been attributed to the presence of functional groups facilitating hydrogen shift.14 For instance, for 4-chlorobenzaldehyde it had been estimated that ca. 1–2% of the NH-nitrone should be present in equilibrium.15 Radom and co-workers studied computationally nitrosomethane and its nitrone and oxime isomers.16 These authors concluded that NH-nitrones should be observable even though a barrier of 70.8 kcal mol−1 (MP3/6-31G++) was estimated for the 1,2-H shift between formaldoxime and formaldonitrone. A further computational study on the oxime–nitrone tautomerism was carried out by Lammertsma and co-workers,17 who demonstrated that the oxime–nitrone tautomerism is thermodynamically unfavoured. More recently, Sousa and co-workers have suggested that the population of the nitrone tautomer should be insignificant on the basis of more accurate calculations.18 However, these recent studies were only concerned with the stability of the different tautomers as well as solvent effects.
The oxime–nitrone tautomerism has important implications in reactivity. When an oxime reacts with an alkene (that can act as both dipolarophile and Michael acceptor if Z is an electron-withdrawing group) three products can be obtained, even though there are four possible reaction pathways (Scheme 2).
 | ||
| Scheme 2 Possible reaction pathways between oximes and electron-poor alkenes. | ||
When oximes react with electron-poor alkenes (Z = EWG), in the absence of any base, the usually preferred pathway is an ene-like reaction (formal Michael addition) via the nitrogen lone pair to yield N-alkylnitrones (Scheme 2, path A).6a The mechanism is expected to be concerted (through TSA) with concomitant nucleophilic attack of the nitrogen atom and H-transfer. In fact, the concertedness of the reaction has already been theoretically demonstrated for related additions of hydroxylamines to electron-poor alkenes.19 Usually, the in situ formed nitrones 3 react with a second molecule of alkene to give the corresponding dipolar cycloaddition (not shown in Scheme 2). On the contrary, when the alkene is unactivated, path D is preferred. As mentioned above, the cycloaddition reaction of the nitrone tautomer to give isoxazolidines 5 (Scheme 2, path D), through a typical concerted transition state TSD, has also been reported in several instances.4 In the case of intramolecular reactions a high dependence of the stereochemistry of the oxime has been observed; whereas E-isomers of oximes react through path A, Z-isomers prefer path D via tautomeric nitrone.13c
Under basic conditions it is also possible to carry out the O-alkylation of the oxime through a typical oxa-Michael reaction.20 However, in the absence of a base the mechanism should take place through a disfavored (due to ring strain) transition state (TSB) leading to O-alkylated derivatives 4 (Scheme 2, path B). To date no evidences have been found of a fourth pathway C consisting of the formation of oxime ethers by nucleophilic attack of the nitrone oxygen, after isomerization of oxime into nitrone. Only two reports described the obtention of O-alkylated oximes by reaction with electron-poor alkenes in the absence of a base.4i,21 By analogy to path A (TSA) the mechanism could be expected to be concerted through TSC. Although it cannot be considered an ene-like reaction because the hydrogen is not allylic, the process involves a 1,4-H shift as in the case of path A.
As part of our ongoing interest in mechanistic aspects of nitrone reactivity,22 we sought to investigate the nitrone–oxime isomerization by computational DFT methods and explore the possibility of a participation of the nitrone tautomer in nucleophilic addition reactions in the absence of a base. We chose for the study the Michael addition of arylamidoximes to 1,2-diaza-1,3-dienes, studied experimentally by Santeusanio and co-workers (Scheme 3).21 This reaction, carried out in the absence of a base, is a rare example of addition of oximes to electron-poor olefins in which O-alkylated oximes 4 are preferentially formed instead nitrones 3.
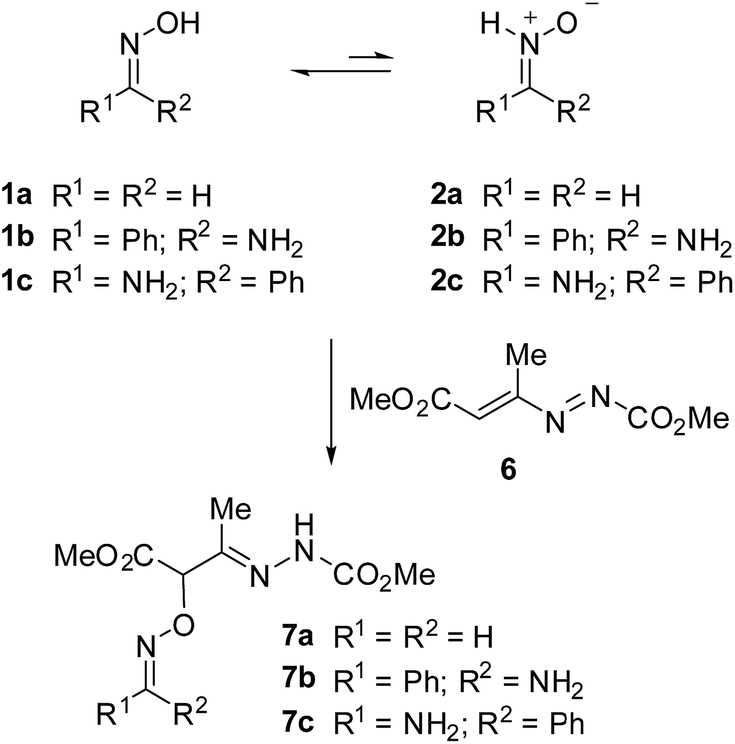 | ||
| Scheme 3 Nucleophilic addition of oximes to 1,2-diaza-1,3-dienes. | ||
Under these circumstances, paths B and C could be competitive, the tautomerism prior the addition being an option. Therefore, aiming to obtain a more accurate description of the oxime–nitrone tautomerism and its implication on nucleophilic addition reactions, this paper presents proofs for a bimolecular isomerization mechanism and, for the first time, an unequivocal characterization of transition state structures in solution involving nitrone tautomers in a nucleophilic addition reaction (Scheme 2, path C).
Computational methods
All of the calculations were performed using the Gaussian09 program.23 Molecular geometries were optimized with the M06-2X functional24 in conjunction with cc-pVTZ basis set.25 This method has been recently used successfully in theoretical investigations with nitrones.22a–f Analytical second derivatives of the energy were calculated to classify the nature of every stationary point, to determine the harmonic vibrational frequencies, and to provide zero-point vibrational energy corrections. The thermal and entropic contributions to the free energies were also obtained from the vibrational frequency calculations, using the unscaled frequencies. All transition structures were characterized by one imaginary frequency. All the located TSs were confirmed to connect to reactants and products by intrinsic reaction coordinate (IRC) analyses.26 The IRC paths were traced using the second order González–Schlegel integration method.27 Calculations have been carried out considering solvent effects (EtOH) with the PCM.28We have considered two models for the study of oxime–nitrone isomerization: (i) the simplest oxime 1a and nitrone 2a, and (ii) the real system studied experimentally,22 consisting of oxime (Z)-1b and nitrone (Z)-2b. For the purpose of comparison, we have also included in the study the (E)-isomers 1c and 2c (Scheme 3).
Results and discussion
Starting materials
We first computed 1a and both E/Z isomers of oximes 1b, c and nitrones 2b, c; since the reaction was carried out in EtOH, we chose this solvent for our studies. In the case of oximes, the anti conformer was the most stable; in fact, the corresponding syn oximes could not be located because all calculations converged to the most stable anti oximes. The optimized geometries of compounds 1a–c and 2a–c are given in Fig. 1.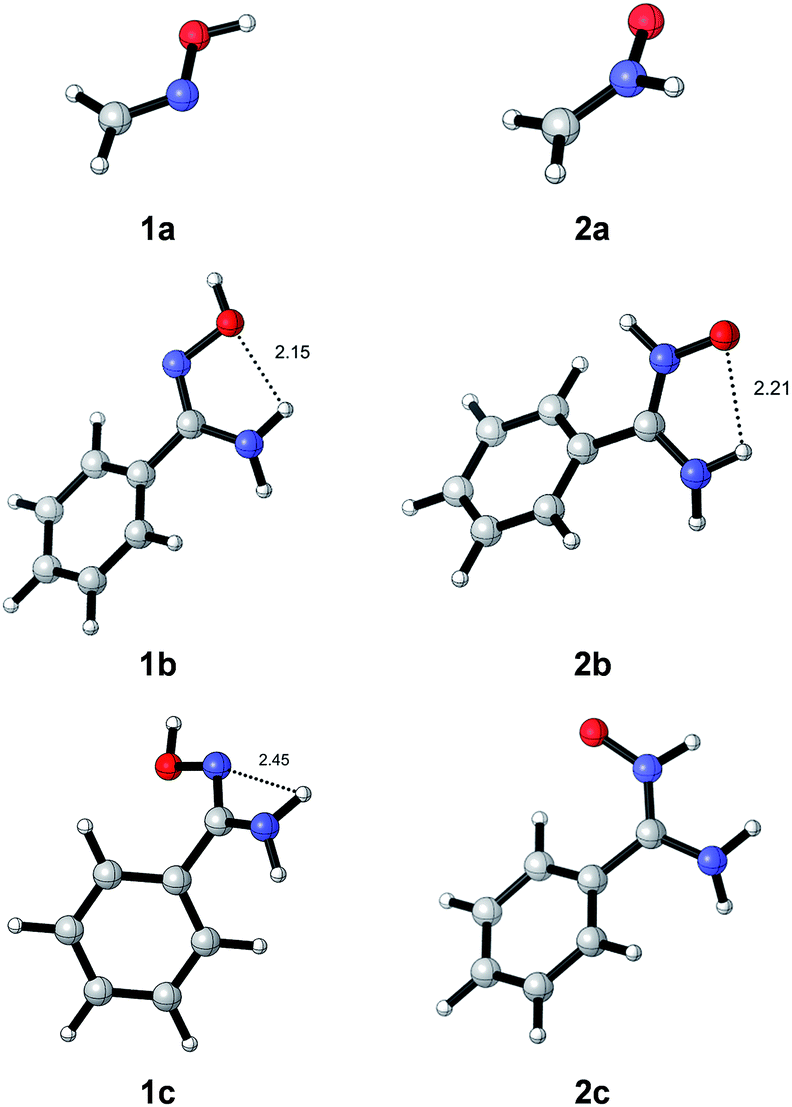 | ||
| Fig. 1 Optimized transition structures (M06-2X/cc-pVTZ/PCM = EtOH) of oximes 1a–c and nitrones 2a–c. Distances are given in angstrom. | ||
Calculations predict the Z-isomer to be more stable than the E-isomer. In the case of oximes, isomer (Z)-1b is 5.8 kcal mol−1 more stable than (E)-1c; for nitrones (Z)-2b is 6.9 kcal mol−1 more stable than (E)-2c. Both (Z)-isomers present a hydrogen bond between the amino group and the oxygen atom (d = 2.15 Å and 2.21 Å for 1b and 2b, respectively). A weaker interaction (d = 2.45 Å) is also observed for (E)-1c between the amino group and the oxime nitrogen. All Z and E isomers 1b, c and 2b, c deviate the phenyl ring from planarity because of the steric hindrance of the phenyl ring. Larger torsion angles are present for (E)-isomers than for (Z)-isomers (CAr2–CAr1–Cox–Nox: 53.8° for (E)-1cvs. 27.5° for (Z)-1b. CAr2–CAr1–Cnit–Nnit: 35.0° for (E)-2cvs. 26.4° for (Z)-2b). When compared oxime 1b and nitrone 2b, the former resulted to be more stable by 9.2 kcal mol−1 in agreement with experimental observations. The E/Z isomerization of oximes and nitrones has been studied in previous reports and it is well document that it can take place through energy barriers of ca. 10 kcal mol−1 for oximes29 and 25 kcal mol−1 for nitrones.21b
Oxime–nitrone tautomerism
Grigg and co-workers initially suggested that 1,2-prototropy in oximes should be a facile process.30 On the other hand, the same authors also mentioned that it is a high energy process when compared with other reactions such as formal Michael additions (actually, ene-like reactions),6a thus explaining the observed behavior in the presence of electron-poor olefins (Scheme 2, path A). With other olefins the isomerization is well demonstrated by trapping the NH-nitrone through a dipolar cycloaddition (Scheme 2, path D).4 Since the early proposal of the 1,2-prototropic route by Grigg and co-workers,3 all authors have assumed this rationale11 in spite of such isomerization mechanism had been discarded by Radom and co-workers 30 years ago.16 In fact, that study considered location of transition structure TS1a for the 1,2-H shift between 1a and 2a (Scheme 4) and assigned a barrier to the process of 70.8 kcal mol−1.16 We also located TS1a in gas phase (Table 1) and considering ethanol as solvent at our standard level of theory (M06-2X/cc-pVTZ). The results for the reaction in EtOH are summarized in Table 2. The geometry of TS1a is given in Fig. 2.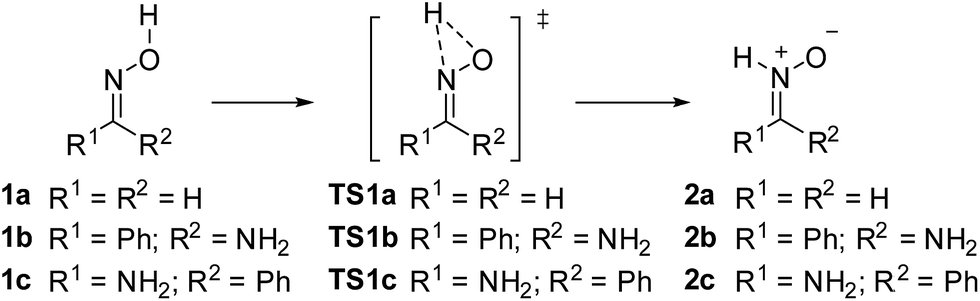 | ||
| Scheme 4 1,2-Shift mechanism of oxime–nitrone isomerization. | ||
 | ||
| Fig. 2 Optimized transition structures (M06-2X/cc-pVTZ/PCM = EtOH) corresponding to 1,2-H shift between oximes 1 and nitrones 2. Distances are given in angstrom. | ||
The calculated barrier for the isomerization in gas phase was 54.7 kcal mol−1. The observed difference of ca. 20 kcal mol−1 with the value obtained by Radom and co-workers in the 80's,16 can be attributed to the use of M06-2X functional which slightly underestimate activation parameters.31 In any case the obtained results give a good illustration of the reaction mechanism pointing out that the process would require a high energy to take place.
When considering solvent effects (EtOH) the barrier for TS1a (53.2 kcal mol−1) is similar to that found in gas phase. The corresponding TS1b starting from 1b to form 2b was also located and a barrier of 49.5 kcal mol−1 was found (Table 2). The transition structure (TS1c) corresponding to the transformation of less stable 1c to form 2c was also located and a higher barrier (56.4 kcal mol−1) was obtained for the process. In any case, high energies (ca. 50 kcal mol−1) are required by the process to take place.
The geometries of the transition structures are given in Fig. 2. Similar distances for hydrogen shift are found in all cases. The N–H distances (1.11–1.13 Å) are shorter than the O–H distances (1.28–1.31 Å). The 1,2-H shift takes place in the plane of the oxime for TS1a and TS1c (H–O1–N2–C3 dihedral angle of 180.0° and 174.7° for TS1a and TS1c, respectively). On the other hand for TS1b the 1,2-H shift takes place out of the plane of the oxime (H–O1–N2–C3 dihedral angle of 132°). This deviation is due to the existing H-bond interaction between an oxygen lone pair and the amino group, which is not present in TS1c. Whereas TS1a is, obviously, completely planar, in TS1b and TS1c the phenyl ring is out the plane formed by the nitrone and amino moieties (by 25° and 43° in TS1b and TS1c, respectively).
It has been suggested that neighboring groups at the periphery of the oxime could promote a more favorable 1,5-H shift in detriment of the assumed 1,2-H shift. This possibility has been exemplified with monoximes of 1,2,3-triketones,32 alk-2-enylamino-substituted pyrans12 and pyrido[1,2-a]pyrimidines.13a,14 In the case of oxime 1b we also explored a similar option by considering the intramolecular protonation of the amino group and further 1,3-H sigmatropic shift (Scheme 5).
 | ||
| Scheme 5 1,2-H shift promoted by neighboring groups. Relative energies (M06-2X/cc-pVTZ/PCM = EtOH) are given in kcal mol−1, between brackets. | ||
According to the calculated mechanism, illustrated in Scheme 5, the isomerization should take place starting from oxime 1b. The initial H-transfer from oxyme to α-amino group takes place along the reaction path in an early stage without formation of any stationary point. The first transition state found in the reaction path corresponds to rotation of the C–N bond. The IRC showed a temporary pyramidalization of C atom with concomitant transformation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O− system into a −C–NO one during the rotation (see ESI†). This rotation is required to achieve the correct orientation of the nitrogen lone pair to receive the proton from the amino group in the second step. In fact, such geometry is that found in intermediate 8 whose zwitterionic nature was confirmed by its large dipole moment (11.6 D). The formation of intermediate 8 is the rate-limiting step. Further evolution of 8 takes place through a 1,3-H shift (TS2b), to yield nitrone 2c (for a complete analysis of the reaction including detailed IRCs and geometries of stationary points see ESI†). The first barrier (79.4 kcal mol−1) corresponding to TS2a is too high to consider this mechanism as a possibility.
N–O− system into a −C–NO one during the rotation (see ESI†). This rotation is required to achieve the correct orientation of the nitrogen lone pair to receive the proton from the amino group in the second step. In fact, such geometry is that found in intermediate 8 whose zwitterionic nature was confirmed by its large dipole moment (11.6 D). The formation of intermediate 8 is the rate-limiting step. Further evolution of 8 takes place through a 1,3-H shift (TS2b), to yield nitrone 2c (for a complete analysis of the reaction including detailed IRCs and geometries of stationary points see ESI†). The first barrier (79.4 kcal mol−1) corresponding to TS2a is too high to consider this mechanism as a possibility.
Next, we considered the possibility of bimolecular mechanisms. In the presence of a protic solvent as ethanol, a solvent molecule can transfer a proton to the nitrogen atom while receiving a different one from the oxime moiety, according to the mechanism illustrated in Scheme 6.
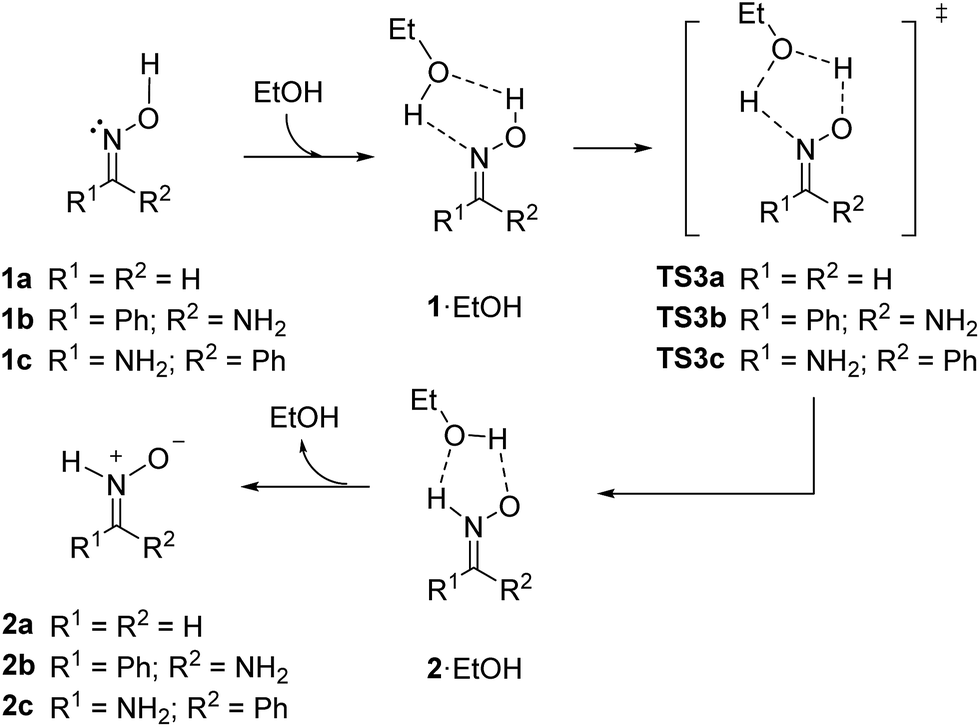 | ||
| Scheme 6 Ethanol-catalyzed bimolecular isomerization of oxime 1 into nitrone 2 (M06-2X/cc-pVTZ/PCM = EtOH). | ||
Similar results were obtained for the model system (series a) and for the real ones (series b and c). After formation of an initial complex between the oxime and a molecule of ethanol, promoted by H-bonding as depicted in Scheme 6, exchange of protons through TS3 give rise to a new complex between ethanol and nitrone leading to isomerized nitrone 2. The corresponding transition structures and the initial and final complexes formed with ethanol were calculated. The results are summarized in Table 3.
| ΔE0a | ΔGa | |
|---|---|---|
| a Referred to 1a or 1b; a discrete molecule of EtOH (ΔE0 = −154.950425 hartrees; ΔG = −154.975812 hartrees) has been considered when necessary. | ||
| 1a | 0.0 | 0.0 |
| 1a·EtOH | −5.5 | 4.1 |
| TS3a | 16.1 | 27.1 |
| 2a·EtOH | 3.0 | 12.5 |
| 2a | 9.8 | 10.0 |
| 1b | 0.0 | 0.0 |
| 1b·EtOH | −6.5 | 3.4 |
| TS3b | 13.2 | 24.5 |
| 2b·EtOH | −0.1 | 10.0 |
| 2b | 9.1 | 9.2 |
| 1c | 6.2 | 5.8 |
| 1c·EtOH | −0.8 | 9.1 |
| TS3c | 19.0 | 30.1 |
| 2c·EtOH | 6.6 | 16.8 |
| 2c | 15.9 | 16.1 |
In the bimolecular process the formation of an initial complex should be stabilized by H-bonding. At the same time, the loss of translational entropy is much higher than in a unimolecular process; consequently, the value of free energy is markedly overestimated.33 Because of it, in order to compare the stability of reagents and products with initial and final complexes, it is more advisable to consider electronic energy (E0) rather than free energy (G) where the entropic factor is introduced.34 According to E0 values, complexes 1·EtOH are ca. 5–7 kcal mol−1 more stable than the corresponding isolated oximes (5.5 kcal mol−1 for 1a, 6.5 kcal mol−1 for 1b and 7.0 kcal mol−1 for 1c). Complexes 2·EtOH are about 7–9 kcal mol−1 more stable than the parent isolated nitrones (6.8 kcal mol−1 for 2a, 9.2 kcal mol−1 for 2b and 9.3 kcal mol−1 for 2c).
Once formed the complex the isomerization takes place in the single entity oxime–EtOH to form the nitrone–EtOH entity, as in a unimolecular reaction, so the relative activation entropy should be close to zero and Gibbs energy can be adequately considered for evaluating the barrier of the process. Once formed the initial complex TS3 controls the rate of the reaction. Similar energy barriers were found in all cases (27.1 kcal mol−1 for the model 1a/2a and 24.5 kcal mol−1 for the most stable Z-isomer (1b/2b) of the real system). The obtained values were 30 kcal mol−1 lower than the unimolecular process discussed above. The geometry of the transition structures TS3a–c showed some differences between the model and the real system (Fig. 3). For TS3a similar distances were found between heteroatoms and hydrogens (1.26–1.28 Å) with the only exception of the shorter OEtOH–H distance of 1.17 Å. In the case of TS3b and TS3c the distance O1–H (1.09 Å in both) was slightly shorter than the N2–H distance (1.12 Å in both). However, the distance between hydrogens and the oxygen atom of the ethanol molecule was much longer (1.41–1.47 Å). The spatial disposition of hydrogen atoms and heteroatoms were rather similar in all transition structures TS3. The higher stability of TS3b over TS3c is due to the extra H-bond interaction between O1 and the amino group.
 | ||
| Fig. 3 Optimized transition structures (M06-2X/cc-pVTZ/PCM = EtOH) corresponding to isomerization of 1 into 2 promoted by a discrete molecule of EtOH. Distances are given in angstrom. | ||
The isomerization also takes place in aprotic solvents and under a variety of conditions. Consequently, it is plausible to consider, independently of the environment, a bimolecular mechanism in which only two molecules of oxime/nitrone are exclusively involved (Scheme 7). Again, formation of initial and final complexes should be considered. The corresponding transition structures and the initial and final complexes were calculated. The results are summarized in Table 4. The geometries of the transition structures are given in Fig. 4 (for geometries of complexes see ESI†).
 | ||
| Scheme 7 Bimolecular mechanism involving exclusively two molecules of oxime/nitrone. | ||
| ΔE0a | ΔGa | |
|---|---|---|
| a Referred to 1a or 1b; two molecules have been considered when necessary. b Barrier calculated from E values, without ZPVE correction. After ZPVE correction the value of the barrier is 16.7 kcal mol−1 revealing the introduction of an experimental error that situates 2c·2c above TS4. This error only affected TS4c since the rest of energy barrier values are similar after ZPVE correction. | ||
| 1a | 0.0 | 0.0 |
| 1a·1a | −8.6 | 1.4 |
| TS4a | 7.6 | 18.7 |
| 2a·2a | 8.2 | 18.1 |
| 2a | 9.8 | 10.0 |
| 1b | 0.0 | 0.0 |
| 1b·1b | −11.1 | 0.0 |
| TS4b | 2.9 | 15.3 |
| 2b·2b | 2.8 | 14.5 |
| 2b | 9.1 | 9.2 |
| 1c | 6.2 | 5.8 |
| 1c·1c | 0.9 | 11.9 |
| TS4c | 20.1b | 28.2 |
| 2c·2c | 17.7 | 28.7 |
| 2c | 15.9 | 16.1 |
 | ||
| Fig. 4 Optimized transition structures (M06-2X/cc-pVTZ/PCM = EtOH) corresponding to isomerization of 1 into 2 through a bimolecular mechanism. Distances are given in angstrom. | ||
By considering E0 values as mentioned above, complexes 1·1 are more stable than the corresponding isolated oximes (8.6 kcal mol−1 for 1a, 11.1 kcal mol−1 for 1b and 5.3 kcal mol−1 for 1c). Complexes 2a·2a and 2c·2c are about 2 kcal mol−1 more stable than the parent isolated nitrones (1.6 and 1.8 kcal mol−1 for 2a and 2c, respectively). A greater difference (6.3 kcal mol−1) was observed between complex 2b·2b and the isolated nitrone 2b. Again, the responsible for this difference is the H-bond interaction present in 2b.
The isomerization from 1a·1a to 2a·2a is endergonic by 16.7 kcal mol−1 with an energy barrier (TS4a) of 18.7 kcal mol−1 from the ground state. In the case of the real system, the formation of two (Z)-nitrones from two molecules of (Z)-oxime is endergonic by 14.5 kcal mol−1. Calculations predicted a barrier (TS4b) of 15.3 kcal mol−1 which is a lower value than those obtained for 1,2-H shift (79.4 kcal mol−1), and intramolecular- and EtOH-assisted isomerizations (24.5 kcal mol−1).35 Similarly, if we consider the less stable (E)-isomers the isomerization from oximes to nitrones is endergonic by 16.8 kcal mol−1 with an energy barrier (TS4c) of 16.4 kcal mol−1.
The transition structure for the model isomerization of 1a into 2a is symmetrical with the hydrogen breaking and forming bonds at distances of 1.19 Å and 1.33 Å from nitrogen and oxygen atoms, respectively. The transition state is completely planar without any deviation out of plane. On the other hand, for the real system the presence of the substituents distort the symmetrical disposition of the transition structure and an asynchronous H-exchange is observed. In fact, for 1b several transition structures displaying minor conformational differences were located. The most stable TS4b and TS4c are given in Fig. 4. The shortest distance (1.10 Å in both TS4b and TS4c) corresponded to one forming N2–H bond, the other one having a distance of 1.26 Å for TS4b and 1.25 Å for TS4c. The longest distances correspond to the breaking O1–H bonds (1.49 Å and 1.50 Å in TS4b and TS4c, respectively) opposed to the above mentioned shortest N2–H forming bonds. The remaining O1–H breaking bonds showed distances of 1.23 Å and 1.25 Å in TS4b and TS4c, respectively. The hydrogen atoms and the heteroatoms involved in the proton exchange are all in the same plane without deviation. The only deviation out of the plane observed corresponded to the phenyl rings, already discussed above for both oximes and nitrones. Again, the difference in stability between TS4b and TS4c is attributed to a cooperative effect of the extra H-bonding interactions in TS4b. Such interactions are stronger (indicated by a shorter distance of 2.17 Å) in the side in which the hydrogen transfer towards the formation of the nitrone is more advanced.
In summary, these data with the simplest oxime 1a and a real system 1b corroborate that the oxime–nitrone isomerization is a bimolecular process involving a hydrogen transfer from an oxime molecule to another. The lowest barrier (15.3 kcal mol−1) for the isomerization has been found for the interconversion of two (Z)-oxime molecules into two (Z)-nitrone molecules; starting from (E)-isomers the barrier was found to be 16.4 kcal mol−1. Higher barriers were found for the other mechanisms investigated including unimolecular 1,2-shift (49.5 kcal mol−1), neighboring group-assisted (79.4 kcal mol−1) mechanisms, and bimolecular ethanol-assisted (24.5 kcal mol−1) isomerization.
Michael addition of arylamidoximes to 1,2-diaza-1,3-dienes
As mentioned above, the reaction between an oxime and an electron-poor olefin, in the absence of a base, proceeds as an ene-like reaction to yield nitrones (Scheme 2, path A). Interestingly, the reaction of oxime 1b with 1,2-diaza-1,3-dienes such as 6, in the absence of any base and in EtOH as a solvent, yields the corresponding O-alkylated oxime 7,21 representing an exception to the usually observed behavior. 1,2-Diaza-1,3-dienes are well-known as Michael acceptors in β-position to the diazo functionality,36 which is the directing electron-withdrawing group. This has been corroborated by reactivity indices calculations.37 In order to investigate whether the nucleophilic addition to 6 takes place via the oxime 1b or the nitrone 2b (after isomerization) we carried out a systematic computational study of the process in which we also included the corresponding (E)-isomers 1c and 2c (for a preliminary study with nitrone 1a see ESI†).1,2-Diaza-1,3-dienes like 6 can adopt s-trans and s-cis conformations involving three dihedral angles. The most stable conformers (by ca. 4–5 kcal mol−1)38 correspond to an s-trans conformation at the central dihedral angle.39 However, the s-cis conformation might lead to stabilizing interactions that could furnish more stable situations. Because of it, both conformations have been taken into consideration. A comprehensive exploration of the PES of the reaction covering a total of 192 initial transition structures was carried out (for details see ESI†).
In principle, both the oxime 1b and nitrone 2b can react with 1,2-diaza-1,3-diene 6 through the corresponding transition structures TS5 and TS6 to provide zwitterions P1 and P2 (Scheme 8). Further migration of the proton, through enol P3, yields the final product 7 observed experimentally. Such a migration might also occur during the formation of the new C–O bond, depending on the interactions found in the corresponding transition structures. Of course, it will be necessary to consider energetics of the isomerization discussed in the preceding section for the full analysis of the process. Also, E/Z isomerization of oxime and/or nitrone tautomer could occur during the reaction. Because of it we have included such variability in the analysis as discussed above (not shown in Scheme 8).
 | ||
| Scheme 8 Mechanisms for the reaction between 1b and 6. Only (Z)-isomers are shown. | ||
The above-mentioned exploratory work of the PES for the nucleophilic attack provided, after full optimization at M06-2X/cc-pVDZ/PCM = EtOH level, 99 independent transition structures from which only 27 account for 100% of probability. Moreover, from those 27, only 8 contribute with more than 1%, accounting for 97.4% of probability. Notably, all TSs in the first 54 positions correspond to a nucleophilic attack of the nitrone tautomer, the first 17 corresponding to (Z)-2b. The first instance of a transition structure corresponding to the nucleophilic attack of the oxime tautomer appears at 55th position with a relative free energy of 10.9 kcal mol−1 (for details see ESI†). Next, in order to obtain more accurate energy barriers for the reaction, we considered transition structures up to 4 kcal mol−1 of relative difference (19 TSs accounting for 99.8%) and they were full optimized at the highest triple-ζ level M06-2X/cc-pVTZ/PCM = EtOH. The results are given in Table 5. As mentioned above all transition structures correspond to the attack of nitrone (Z)-2b (Table 5, entries 1–18) with one exception that corresponds to (E)-2c (Table 5, entry 19), the most stable transition structure for the (E)-series. In order to compare the barriers of the different reaction pathways, we also included in triple-ζ optimization the most stable cases for the addition of oximes (Z)-1b and (E)-1c (Table 5, entries 20 and 21, respectively).40
| Entry | Taut.b | TS | Facec | Stag.d | αe | βe | γe | ΔE0f | ΔGf | Rel. ab.g (%) | Im. freq. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Only those below 4 kcal mol−1 of relative free energy in the 2ζ study have been considered for the triple-ζ one (see text). b Referred to the tautomer that reacts with 6. c Indicates the face of the oxime/nitrone. d Three staggered orientations of the oxime/nitrone attack to 6, defined as “d” (droite), “g” (gauche) and “a” (anti); see ESI for definition of d/g/a. e “c” and “t” correspond to s-cis and s-trans conformations of the considered dihedral angles of 6. f With respect to the most stable one. g According to Boltzmann's distribution. | |||||||||||
| 1 | 2b | TS6a | Re | g | t | c | c | 0.0 | 0.0 | 23.4 | −317.1 |
| 2 | TS6b | Re | g | t | t | c | 0.5 | 0.1 | 20.2 | −304.1 | |
| 3 | TS6c | Re | g | t | c | t | 0.4 | 0.1 | 20.2 | −301.1 | |
| 4 | TS6d | Si | d | t | c | c | 1.6 | 0.3 | 15.0 | −331.7 | |
| 5 | TS6e | Si | g | t | c | c | 1.4 | 0.6 | 8.6 | −313.4 | |
| 6 | TS6f | Si | d | t | t | c | 2.5 | 0.9 | 4.8 | −321.6 | |
| 7 | TS6g | Si | g | t | c | t | 2.4 | 1.5 | 1.9 | −313.8 | |
| 8 | TS6h | Si | d | t | c | t | 2.9 | 1.6 | 1.7 | −330.8 | |
| 9 | TS6i | Re | a | t | c | c | 3.3 | 1.8 | 1.1 | −276.5 | |
| 10 | TS6j | Re | g | t | t | t | 1.8 | 1.9 | 0.9 | −297.2 | |
| 11 | TS6k | Re | g | c | c | c | 4.0 | 2.0 | 0.8 | −291.0 | |
| 12 | TS6l | Si | d | t | t | t | 3.4 | 2.2 | 0.6 | −317.3 | |
| 13 | TS6m | Si | g | t | t | t | 3.7 | 2.6 | 0.3 | −318.4 | |
| 14 | TS6n | Re | a | t | t | c | 4.2 | 2.7 | 0.3 | −277.4 | |
| 15 | TS6o | Si | d | c | c | c | 4.3 | 3.4 | 0.1 | −321.6 | |
| 16 | TS6p | Re | g | c | c | t | 5.1 | 3.8 | 0.0 | −296.0 | |
| 17 | TS6q | Si | d | c | t | c | 4.9 | 4.0 | 0.0 | −321.4 | |
| 18 | TS6r | Si | d | c | c | t | 5.1 | 4.0 | 0.0 | −323.2 | |
| 19 | 2c | TS6s | Re | g | t | c | c | 5.1 | 5.0 | 0.0 | −254.6 |
| 20 | 1b | TS5a | Re | a | t | c | c | 14.2 | 13.2 | 0.0 | −386.4 |
| 21 | 1c | TS5b | Re | a | c | t | c | 19.5 | 19.4 | 0.0 | −552.8 |
The three most stable transition states TS6a–c (within a range of 0.1 kcal mol−1) and TS6j accounting for 64.7% correspond to the same approach; they only differ on variations at β and γ dihedral angles of the 1,2-diaza-1,3-diene 6. The same situation is found for TS6d, f, h and TS6e, g, TS6i representing a different approach. In consequence, it is possible to limit the discussion by considering only 4 models, namely TS6a, TS6d, TS6e and TS6i that adequately represent 10 situations in a range of 2.0 kcal mol−1 of relative energy, accounting for 97.9% of Bolztmann's distribution. The rest of TSs, i.e.TS6k–s, are at least 2.0 kcal mol−1 above the most stable TS6a and account for the remaining 2.1%. The geometries of TS6a, TS6d, TS6e and TS6i are given in Fig. 5. Whereas in TS6a and TS6i the attack is carried out from the Re face of the nitrone, in TS6d and TS6e, the attack comes from the Si face of the nitrone. The forming bond distances are similar in all cases. TS6a and TS6e correspond to droite approaches with N2–O1–C1′–C2′ angles of 44.1° and 13.0°, respectively. While T6d corresponds to a gauche approach, TS6i corresponds to an anti approach.
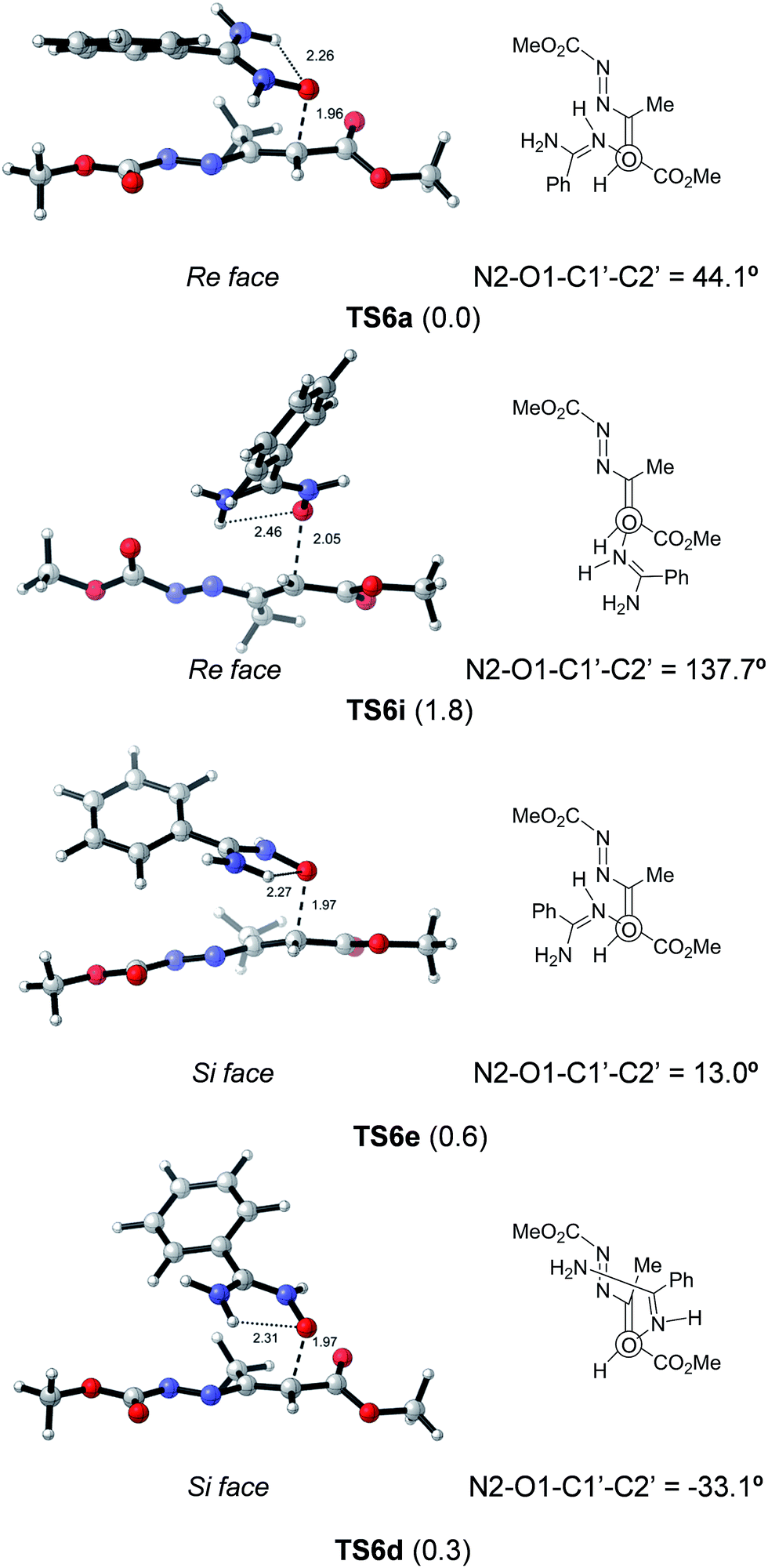 | ||
| Fig. 5 Selected optimized transition structures (M06-2X/cc-pVTZ/PCM = EtOH) corresponding to the nucleophilic attack of 2b to 6. The more stable TSs for each approach are showed. Relative energies are given in kcal mol−1. Distances are given in angstroms. | ||
The four models illustrated in Fig. 5 are in a range of 1.8 kcal mol−1, close to experimental error so, any of them could be considered valid. In any case, the favored addition through the (Z)-nitrone tautomer is fully confirmed. The addition of (E)-nitrone 2c was found to be 5.0 kcal mol−1 higher in energy. The most stable TSs, TS5a and TS5b, corresponding to the addition of oxime tautomers gave even higher energy values (by 13.2 and 19.4 kcal mol−1 for (Z)-oxime 1b and (E)-oxime 1c, respectively).
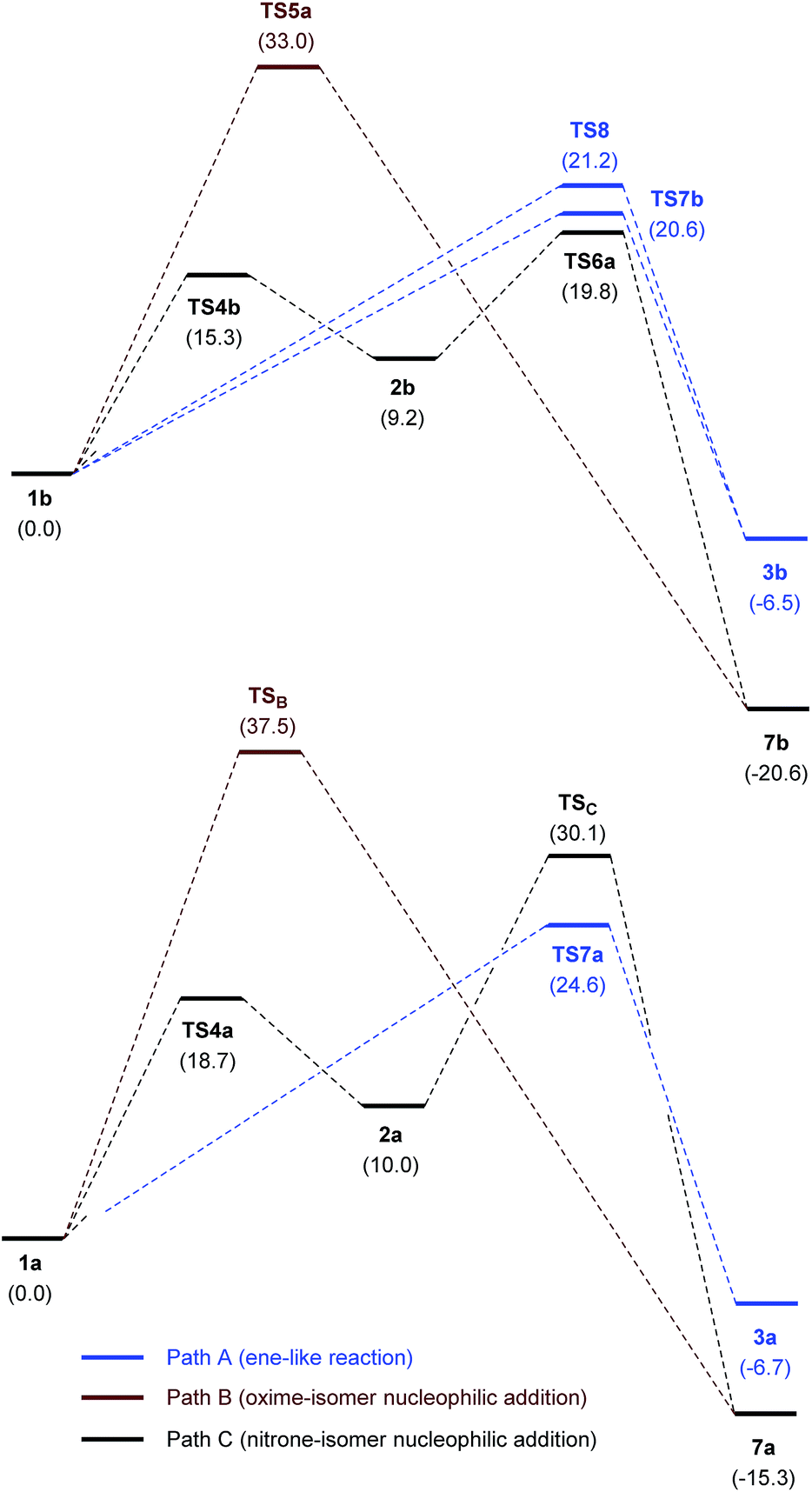
Fig. 6 illustrates the compared simplified energy diagrams for the different isomers showing the energy barriers for the reaction of each isomer. Different final points (not shown in Fig. 6) were obtained for each reaction path; after tautomeric processes those points led to either 7b or 7c, the most stable tautomers (for complete energy diagrams, transition structures and full reaction paths see ESI†).
 | ||
| Fig. 6 Simplified free energy diagrams (M06-2X/cc-PVTZ/PCM = EtOH) for the nucleophilic addition of oximes 1b, c and nitrones 2b, c to 1,2-diaza-1,3-diene 6. TS5a and TS5b correspond to (Z)- and (E)-oximes, respectively. TS6a and TS6b correspond to (Z)- and (E)-nitrones, respectively. | ||
To sum up, even though isomerization of oxime 1b to nitrone 2b has a barrier of 15.3 kcal mol−1 and requires a cost in energy of 9.2 kcal mol−1, the most stable transition state is that corresponding to the attack of the nitrone tautomer through TS6a. According to the Curtin–Hammett principle41 this is the preferred path for the reaction, revealing a rare case in which a nitrone tautomer participates in a nucleophilic addition reaction.
Ene-like reactions and alternative processes
As we discussed in the introduction, ene-like reaction between 1b and 6 (path A, Scheme 2) and dipolar cycloaddition between 2b and 6 (path D, Scheme 2), could be competitive with the nucleophilic Michael addition. In order to validate our model and further confirm the preference by the nitrone-mode addition, we also calculated those alternative paths of the reaction.The possibility of an ene-like reaction (Scheme 2, path A) was first explored. A typical transition structure TSA could be located in a simple model like addition of 1b to dimethyl 2-methylfumarate.42 The IRC analysis corroborated that the reaction was concerted and transfer of hydrogen takes place giving rise to the corresponding nitrone (see ESI†).
On the other hand, the reaction with 1,2-diaza-1,3-diene 6 led to dramatic changes on the located transition structures. A full exploration of the PES corresponding to the nucleophilic attack of the oxime nitrogen was carried out by considering 1b and 1c as well as s-trans-6 and s-cis-6 (for details see ESI†).
No transition structures corresponding to an H-transfer to the carbon atom through a concerted ene-like process could be located; instead, in all cases the hydrogen atom was oriented to the formation of an H-bond either with one of the nitrogen atoms of the diazo group or the carbonyl ester group directly linked to the double bond. The most stable paths were those showed in Scheme 9. TS7b was 0.6 kcal mol−1 lower in energy than TS8 (energy barriers: 20.6 and 21.2 kcal mol−1 for TS7b and TS8, respectively) so, both transition structures can be considered competitive. Transition structure TS7b does not correspond to an ene-like process and the IRC analysis confirmed a partial hydrogen transfer. The resulting zwitterion P4b is assumed to be transformed into the neutral nitrone 9b through a typical tautomeric process. In the case of s-cis-6, IRC of TS8 reflects a complete transfer of hydrogen to the diazo group, leading directly to the neutral nitrone. Any attempt of locating an ene-like transition structure similar to that located for the model methyl 2-methylfumarate, led to one of the TSs located previously during PES exploration (see ESI†).
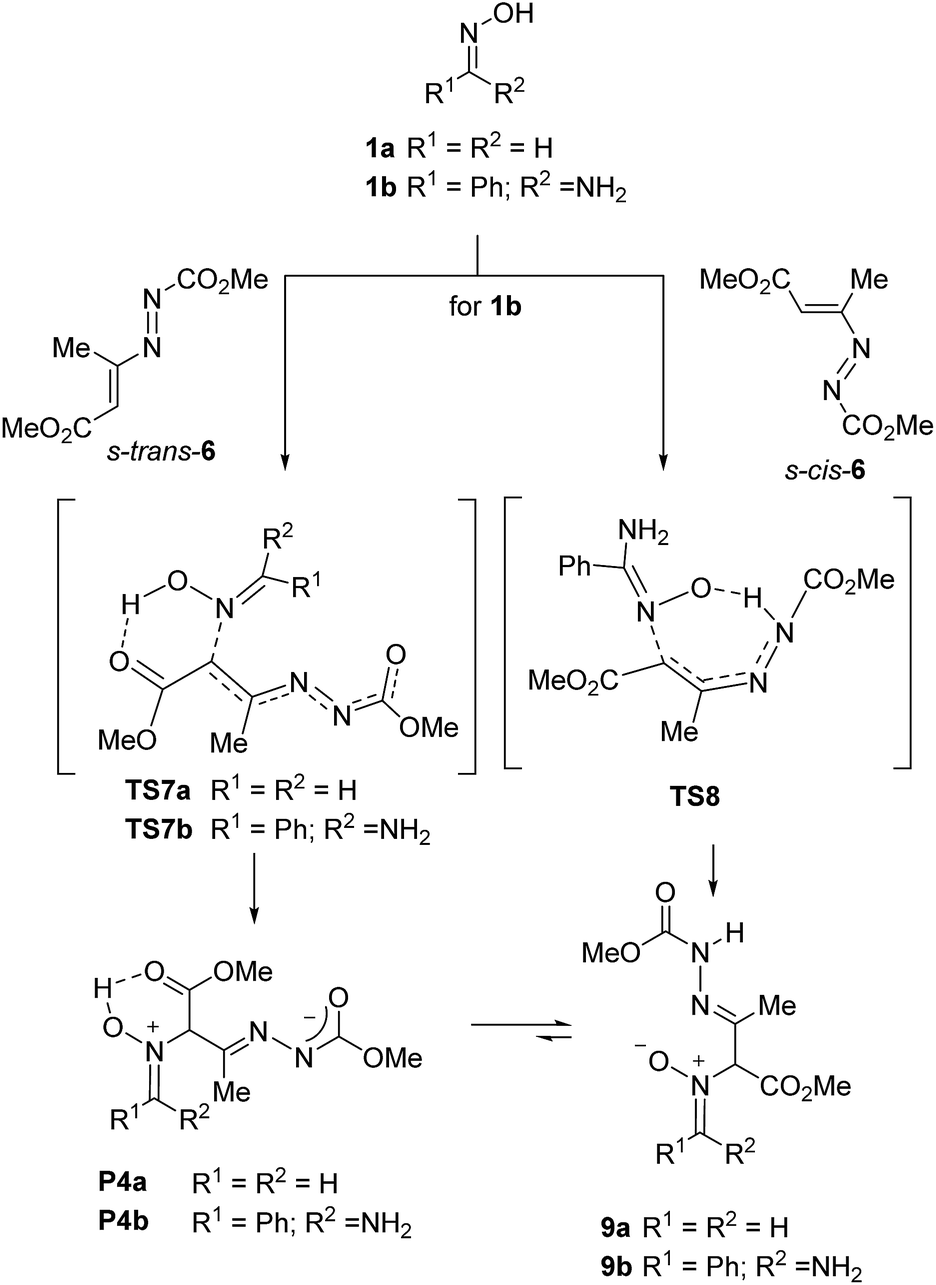 | ||
| Scheme 9 Preferred path for the nucleophilic attack of the oxime nitrogen. | ||
The free energy barriers found for TS7b, and TS8 were 0.8 and 1.4 kcal mol−1, respectively, higher in energy than the most stable TS6a (Fig. 7). These values indicate that the nucleophilic addition is preferred in agreement with experimental observations. However, it should also be considered that both processes might be competitive since the differences observed between barrier values are within the error limit of the calculations. Nevertheless, it would not be surprising that a close inspection of the reaction mixture could reveal the obtention of minor amounts of nitrone 9b.
 | ||
| Fig. 7 Comparative free energy diagrams (M06-2X/cc-pVTZ/PCM = EtOH) for the reaction between 1b and 6 (top) and 1a and 6 (bottom). Relative free energies are given in kcal mol−1. | ||
When the same study was made with the unsubstituted oxime 1a, the nucleophilic addition of the oxime nitrogen leading to the nitrone through TS7a, clearly resulted the most stable situation (5.5 kcal mol−1 lower in energy than the corresponding transition structure TSC; see ESI†) in agreement with previous experimental findings with aldoximes and revealing the particularity of oxime 1b (Fig. 7). These results allow predicting that, in general and in the absence of a base, the ene-like process (Scheme 2, path A) should be preferred for the reaction of an oxime and an electron-poor alkene, but when steric hindrance is present as in the case of ketoximes like 1b, the O-alkylation is a competitive process taking place through the attack of the nitrone tautomer (Scheme 2, path C).
The reason of the different results obtained with 1a and 1b can be found in the different spatial requirements of both oximes. The geometries of transition structures TS7b, TS8 and TS7a are given in Fig. 8. The unsubstituted oxime 1a attacks with a correct orientation of the nitrogen lone pair, through a completely perpendicular eclipsed orientation (O1–N2–C3–C4 dihedral angle of −164.9°) following a typical Burgi–Dunitz trajectory (N2–C3–C4 angle of 108.6°). The perpendicularity of the oxime with respect to the 1,2-diaza-1,3-diene is evidenced by the angle of 98.4° formed between the reactant planes defined by the oxime and the diazadiene (Fig. 8).43 On the other hand, oxime 1b is forced to modify such orientation in order to maintain the Burgi–Dunitz trajectory (N2–C3–C4 angles of 109.0° and 109.5° for TS7b and TS8, respectively) and, at the same time, to avoid unfavorable steric interactions. As a consequence, the nitrogen lone pair is not perfectly aligned with the carbon atom and the situation increases the energy of the corresponding transition structures. The distorsion during the attack of 1b is revealed by the steeper angles formed between the planes defined by the oxime and the diazadiene (57.8° and 62.6° for TS7b and TS8, respectively) with respect to that observed for 1a.
 | ||
| Fig. 8 Optimized transition structures (M06-2X/cc-pVTZ/PCM = EtOH) of TS7b and TS8 corresponding to the ene-like reaction between oxime 1b and 1,2-diaza-1,3-diene 6, and TS7a corresponding to the reaction between 1a and 6. Distances are given in angstrom. | ||
Finally, no concerted transition structures corresponding to path D (Scheme 2) could be located. Moreover, any attempt of locating such concerted transition structures led to some of the already characterized TSs listed in Table 5. In particular, a systematic search and optimization for initial 3,5-endo and 3,5-exo transition structures led to TS6d and TS6a, respectively.
Conclusions
The oxime–nitrone isomerization has been studied computationally and it has been determined that it takes place through a bimolecular mechanism involving two molecules of oxime (or nitrone). The commonly accepted 1,2-H shift can be completely discarded, even if favored by neighboring groups, due to the high energies of the corresponding transition structures. Oximes usually react with electron deficient olefins to give the corresponding nitrones through N-alkylation (formal Michael addition) and concomitant hydrogen transfer. The mechanism of the reaction had been proposed to be an ene-like concerted process and it has been confirmed in the case of a simple model.The O-alkylation observed experimentally in the reaction of oxime 1b to 1,2-diaza-1,3-diene 6, in the absence of a base, can be considered an exception. The computational study carried out on this process has demonstrated that the reaction takes place through the nucleophilic attack of the nitrone tautomer. The addition represents the first example in which the nitrone tautomer is involved into a nucleophilic addition of an oxime. Nevertheless, the N-alkylation should be considered a competitive process. Indeed, it can also be predicted that O-alkylation (though nitrone tautomer) will take place particularly with sterically demanding substrates such as ketoximes like the studied 1b. In the case of substrates that allow a favorable orthogonal approach of the oxime to the electron deficient alkene, the N-alkylation leading to the corresponding nitrone will be preferred.
Acknowledgements
This work was supported by the Spanish Ministerio de Economía y Competitividad (MINECO) (project number CTQ2013-44367-C2-1-P), by the Fondos Europeos para el Desarrollo Regional (FEDER) and the Gobierno de Aragón (Zaragoza, Spain, Bioorganic Chemistry Group, E-10). The authors acknowledge the Institute of Biocomputation and Physics of Complex Systems (BIFI) at the University of Zaragoza for computer time at clusters Terminus and Memento. D. R.-L. thanks the Spanish Ministry of Education (MEC) for a pre-doctoral grant (FPU program).Notes and references
- P. C. Vijfhuizen and J. K. Terlouw, Org. Mass Spectrom., 1977, 12, 63 CrossRef CAS.
- K. J. Dignam and A. F. Hegarty, J. Chem. Soc., Perkin Trans. 2, 1979, 1437 RSC.
- (a) R. Grigg, H. Q. N. Gunaratne and J. Kemp, J. Chem. Soc., Perkin Trans. 1, 1984, 41 RSC; (b) R. Grigg, M. Jordan, A. Tangthongkum, F. W. B. Einstein and T. Jones, J. Chem. Soc., Perkin Trans. 1, 1984, 47 RSC; (c) R. Grigg, Chem. Soc. Rev., 1987, 16, 89 RSC.
- (a) M. Noguchi, S. Nishimura, H. Fujii and A. Kakehi, Heterocycl. Commun., 1999, 5, 133 CrossRef CAS; (b) S. Moutel and M. Shipman, Synlett, 1998, 1333 CrossRef CAS; (c) C. A. D. Sousa, M. L. C. Vale, X. Garcia-Mera and J. E. Rodriguez-Borges, Tetrahedron, 2012, 68, 1682 CrossRef CAS; (d) P. Armstrong, R. Grigg and W. J. Warnock, J. Chem. Soc., Chem. Commun., 1987, 1325 RSC; (e) U. Chiacchio, A. Corsaro, D. Iannazzo, A. Piperno, V. Pistara, A. Rescifina, R. Romeo, G. Sindona and G. Romeo, Tetrahedron: Asymmetry, 2003, 14, 2717 CrossRef CAS; (f) J. A. Rincon, C. Mateos, P. Garcia-Losada and D. J. Mergott, Org. Process Res. Dev., 2015, 19, 347 CrossRef CAS; (g) C. A. D. Sousa, J. E. Rodriguez-Borges and X. Garcia-Mera, Tetrahedron Lett., 2014, 55, 4628 CrossRef CAS; (h) E. Frank, Z. Mucsi, M. Szecsi, I. Zupko, J. Woelfling and G. Schneider, New J. Chem., 2010, 34, 2671 RSC; (i) C. W. Lee, J. Y. Park, H. Kim and K.-W. Chi, Bull. Korean Chem. Soc., 2010, 31, 1172–1176 CrossRef CAS.
- (a) A. Hassner and R. Maurya, Tetrahedron Lett., 1989, 30, 5803 CrossRef CAS; (b) A. Hassner, R. Maurya, A. Padwa and W. H. Bullock, J. Org. Chem., 1991, 56, 2775 CrossRef CAS; (c) A. Hassner, R. Maurya, O. Friedman, H. E. Gottlieb, A. Padwa and D. Austin, J. Org. Chem., 1993, 58, 4539 CrossRef CAS; (d) A. Hassner, K. M. L. Rai and W. Dehaen, Synth. Commun., 1994, 24, 1669 CrossRef CAS; (e) E. Falb, Y. Bechor, A. Nudelman, A. Hassner, A. Albeck and H. E. Gottlieb, J. Org. Chem., 1999, 64, 498 CrossRef CAS; (f) G. V. M. Sharma, I. S. Reddy, V. G. Reddy and A. V. R. Rao, Tetrahedron: Asymmetry, 1999, 10, 229 CrossRef CAS; (g) A. Bhattacharjee, S. Datta, P. Chattopadhyay, N. Ghoshal, A. P. Kundu, A. Pal, R. Mukhopadhyay, S. Chowdhury, A. Bhattacharjya and A. Patra, Tetrahedron, 2003, 59, 4623 CrossRef CAS; (h) L. Gottlieb, A. Hassner and H. E. Gottlieb, Synth. Commun., 2000, 30, 2445 CrossRef CAS; (i) L. Doyle and F. Heaney, Tetrahedron, 2010, 66, 7041 CrossRef CAS; (j) F. Heaney, J. Fenlon, C. O'Mahony, P. McArdle and D. Cunningham, Org. Biomol. Chem., 2003, 1, 4302 RSC.
- (a) R. Grigg, J. Markandu, T. Perrior, S. Surendrakumar and W. J. Warnock, Tetrahedron, 1992, 48, 6929 CrossRef CAS; (b) M. Noguchi, S. Matsumoto, M. Shirai and H. Yamamoto, Tetrahedron, 2003, 59, 4123 CrossRef CAS.
- (a) O. Tamura, T. Mitsuya and H. Ishibashi, Chem. Commun., 2002, 1128 RSC; (b) O. Tamura, N. Iyama and H. Ishibashi, J. Org. Chem., 2004, 69, 1475 CrossRef CAS PubMed; (c) O. Tamura, T. Mitsuya, X. Huang, Y. Tsutsumi, S. Hattori and H. Ishibashi, J. Org. Chem., 2005, 70, 10720 CrossRef CAS PubMed.
- (a) O. Tamura, N. Morita, Y. Takano, K. Fukui, I. Okamoto, X. Huang, Y. Tsutsumi and H. Ishibashi, Synlett, 2007, 658 CrossRef CAS; (b) N. Morita, K. Fukui, J. Irikuchi, H. Sato, Y. Takano, I. Okamoto, H. Ishibashi and O. Tamura, J. Org. Chem., 2008, 73, 7164 CrossRef CAS PubMed; (c) N. Morita, R. Kono, K. Fukui, A. Miyazawa, H. Masu, I. Azumaya, S. Ban, Y. Hashimoto, I. Okamoto and O. Tamura, J. Org. Chem., 2015, 80, 4797 CrossRef CAS PubMed; (d) N. Morita, R. Kono, K. Fukui, A. Miyazawa, H. Masu, I. Azumaya, S. Ban, Y. Hashimoto, I. Okamoto and O. Tamura, J. Org. Chem., 2015, 80, 4797 CrossRef CAS PubMed.
- (a) E. Frank, Z. Mucsi, M. Szecsi, I. Zupko, J. Wolflingg and G. Schneider, New J. Chem., 2010, 34, 2671 RSC; (b) A. K. Nacereddine, H. Layeb, F. Chafaa, W. Yahia, A. Djerourou and L. R. Domingo, RSC Adv., 2015, 5, 64098 RSC.
- (a) P. Merino, in Science of Synthesis, ed. E. Schaumann, George Thieme, Stuttgar, 2011, vol. 2010/4, p. 325 Search PubMed; (b) P. Merino, in Science of Synthesis, ed. D. Bellus and A. Padwa, George Thieme, Stuttgart, 2004, vol. 27, p. 511 Search PubMed; (c) R. Matute, S. Garcia-Viñuales, H. Hayes, M. Ghirardello, A. Daru, T. Tejero, I. Delso and P. Merino, Curr. Org. Synth., 2015 DOI:10.2174/1570179412666150914200035.
- (a) M. Noguchi, H. Okada, M. Tanaka, S. Matsumoto, A. Kakehi and H. Yamamoto, Bull. Chem. Soc. Jpn., 2001, 74, 917 CrossRef CAS; (b) M. Noguchi, H. Okada, S. Nishimura, Y. Yamagata, S. Takamura, M. Tanaka, A. Kakehi and H. Yamamoto, J. Chem. Soc., Perkin Trans. 1, 1999, 185 RSC; (c) I. Walton, M. Davis, L. Yang, Y. Zhang, D. Tillman, W. L. Jarrett, M. T. Huggins and K. J. Wallace, Magn. Res. Chem., 2011, 49, 205 CrossRef CAS PubMed; (d) F. Heaney, S. Bourke, D. Cunningham and P. McArdle, J. Chem. Soc., Perkin Trans. 2, 1998, 547 RSC; (e) F. Heaney and S. Bourke, J. Chem. Soc., Perkin Trans. 1, 1998, 955 RSC.
- M. Gotoh, T. Mizui, B. Sun, K. Hirayama and M. Noguchi, J. Chem. Soc., Perkin Trans. 1, 1995, 1857 RSC.
- (a) M. Gotoh, B. Sun, K. Hirayama and M. Noguchi, Tetrahedron, 1996, 52, 887 CrossRef CAS; (b) F. Heaney and C. O'Mahony, J. Chem. Soc., Perkin Trans. 1, 1998, 341 RSC; (c) C. O'Mahony and F. Heaney, Chem. Commun., 1996, 167 RSC.
- M. Shirai, H. Kuwabara, S. Matsumoto, H. Yamamoto, A. Kakehi and M. Noguchi, Tetrahedron, 2003, 59, 4113 CrossRef CAS.
- J. E. Reimann and W. P. Jencks, J. Am. Chem. Soc., 1966, 88, 3973 CrossRef CAS PubMed.
- P. D. Adeney, W. J. Bouma, L. Radom and W. R. Rodwell, J. Am. Chem. Soc., 1980, 102, 4069 CrossRef CAS.
- J. A. Long, N. J. Harris and K. Lammertsma, J. Org. Chem., 2001, 66, 6762 CrossRef CAS PubMed.
- C. A. D. Sousa, M. L. C. Vale, X. Garcia-Mera and J. E. Rodríguez-Borges, Tetrahedron, 2012, 68, 1682 CrossRef CAS.
- (a) A. G. Moglioni, E. Muray, J. A. Castillo, A. Alvarez-Larena, G. Y. Moltrasio, V. Branchadell and R. M. Ortuno, J. Org. Chem., 2002, 67, 2402 CrossRef CAS PubMed; (b) J. Moran, J. Y. Pfeiffer, S. I. Gorelsky and A. M. Beauchemin, Org. Lett., 2009, 11, 1895 CrossRef CAS PubMed.
- (a) H. M. Meshram, B. Eeshwaraiah, M. Sreenivas, D. Aravind, B. S. Sundar and J. S. Yadav, Synth. Commun., 2009, 39, 1857 CrossRef CAS; (b) A. Pohjakallio and P. M. Pihko, Chem.–Eur. J., 2009, 15, 3960 CrossRef CAS PubMed; (c) B. Tan, Z. Shi, P. J. Chua, Y. Li and G. Zhong, Angew. Chem., Int. Ed., 2009, 48, 758 CrossRef CAS PubMed; (d) T. J. Martin, V. G. Vakhshori, Y. S. Tran and O. Kwon, Org. Lett., 2011, 13, 2586 CrossRef CAS PubMed; (e) A. Carlone, G. Bartoli, M. Bosco, F. Pesciaioli, P. Ricci, L. Sambori and P. Melchiorre, Eur. J. Org. Chem., 2007, 5492 CrossRef CAS.
- O. A. Attanasi, L. Cotarca, G. Favi, P. Filippone, F. R. Perrulli and S. Santeusanio, Synlett, 2009, 1583 CAS.
- (a) D. Roca-López, V. Polo, T. Tejero and P. Merino, J. Org. Chem., 2015, 80, 4076 CrossRef PubMed; (b) D. Roca-López, T. Tejero and P. Merino, J. Org. Chem., 2014, 79, 8358 CrossRef PubMed; (c) P. Merino, T. Tejero and A. Diez Martinez, J. Org. Chem., 2014, 79, 2189 CrossRef CAS PubMed; (d) D. Roca-López, V. Polo, T. Tejero and P. Merino, Eur. J. Org. Chem., 2015, 4143 CrossRef; (e) D. Roca-López, T. Tejero, P. Caramella and P. Merino, Org. Biomol. Chem., 2014, 12, 517 RSC; (f) A. Daru, D. Roca-López, T. Tejero and P. Merino, J. Org. Chem., 2016, 81, 673 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- (a) T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef; (b) R. A. Kendall, T. H. Dunning Jr. and R. J. Harris, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS; (c) D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358 CrossRef CAS; (d) E. Papajak, J. Zheng, X. Xu, H. R. Leverentz and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 3027 CrossRef CAS PubMed.
- (a) K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS; (b) K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- (a) C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef; (b) C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853 CrossRef.
- (a) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (b) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS; (d) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS.
- J. Galvez and A. Guirado, J. Comput. Chem., 2010, 31, 520 CAS.
- R. Grigg, F. Heaney, J. Markandu, S. Surendrakumar, M. Thornton-Pett and W. J. Warnock, Tetrahedron, 1991, 47, 4007 CrossRef CAS.
- R. Jasinski, Comput. Theor. Chem., 2014, 1046, 93–98 CrossRef CAS.
- R. Grigg and S. Thianpantangul, J. Chem. Soc., Perkin Trans. 1, 1984, 653 RSC.
- H.-X. Zhou and M. K. Gilson, Chem. Rev., 2009, 109, 4902 Search PubMed.
- The prediction of accurate Gibbs free energy values for medium systems in solution remains a challenge for present-day DFT methods. The absolute errors might be significant for association/dissociation steps; however, for isomeric processes, favorable error cancellation is expected, leading to smaller relative errors as compared to their absolute values.
- Considering that in the experimental conditions ethanol is the solvent, the formation of the oxime pair 1·1 requires desolvation of two molecules of oxime which is not necessary in the ethanol-assisted mechanism. However, it is also necessary to consider the additional solvation energy of the newly formed oxime dimer, which compensate desolvation of oximes 1 (for a detailed analysis of the situation see ESI†).
- (a) S. Mantenuto, F. Mantellini, G. Favi and O. A. Attanasi, Org. Lett., 2015, 17, 2014 CrossRef CAS PubMed; (b) L. De Crescentini, O. A. Attanasi, L. A. Campisi, G. Favi, S. Lillini, F. Ursini and F. Mantellini, Tetrahedron, 2015, 71, 7282 CrossRef CAS; (c) E. Juhasz-Toth, G. Favi, O. A. Attanasi, A. C. Benyei and T. Patonay, Synlett, 2014, 25, 2001 CrossRef CAS.
- R. Majer, O. Konechnaya, I. Delso, T. Tejero, O. A. Attanasi, S. Santeusanio and P. Merino, Org. Biomol. Chem., 2014, 12, 8888 CAS.
- E. M. Brasil, R. S. Borges, O. A. S. Romero, C. N. Alves, J. A. Saez and L. R. Domingo, Tetrahedron, 2012, 68, 6902 CrossRef CAS.
- For a complete conformational analysis of 6 see ESI†.
- Actually, entries 19–21 were selected after optimization of similar structures within a range of 2 kcal mol−1 to ensure that the most stable situations are chosen.
- J. I. Seeman, Chem. Rev., 1983, 83, 83 CrossRef CAS.
- We initially used this model because it represents the exchange of the methoxycarbonyldiazo group by a simple ester group. For details see ESI†.
- The dihedral angle between those planes has been found by: (1) calculating the equation for each plane from cartesian atomic cooordinates of three atoms of oxime and three atoms of 1,2-diaza-1,3-diene, and (2) calculating the corresponding angle determined by the normal vectors of the planes.
Footnote |
| † Electronic supplementary information (ESI) available: Details on calculations corresponding to oxime–nitrone tautomerism including unimolecular 1,2-H shift direct and promoted by neighboring groups; bimolecular mechanisms promoted by EtOH and via oxime dimer complexes. Conformational analysis and reactivity indices of 1,2-diaza-1,3-diene 6. Details on calculations corresponding to Michael addition of oximes 1a and 1b to 6. Details on calculations corresponding to ene-like processes between oxime 1a and 6, and oxime 1b and methyl 2-methylfumarate and 6. Cartesian coordinates of optimized structures. See DOI: 10.1039/c6ra02638a |
| This journal is © The Royal Society of Chemistry 2016 |